From Dr Richard Walding
Updated 20 March 2026
SCIENCE ENQUIRY "INVESTIGATE PROPERTIES OF ACIDS AND BASES" 1. Comparing monotprotic an polyprotic acids by titration 2. Comparing the conductivity of monoprotic and polyprotic acids. 3. How does the conductivity of weak acids vary with concentration? 4. How does the [H+] concentration of acetic acid vary with concentration? 5. How does the pH of acetic acid vary with concentration? 6. How does the pH of water vary with temperature? 7. How does the pH of acetic acid vary with temperature? 8. How acurate are thermometric acid-base titrations • Wine and soft drink titration experiments • Soda water titration experiments
SCIENCE ENQUIRY "INVESTIGATE CONDUCTOMETRIC TITRATIONS"
How does the conductivity of a solution change during an acid-base conductometric titration?
SCIENCE INQUIRY: "INVESTIGATE GALVANIC CELLS" • Open circuit galvanic cell experiments • Closed circuit galvanic cell experiments
SCIENCE INQUIRY: "INVESTIGATE FACTORS THAT AFFECT ELECTROLYSIS" • Electrolysis experiments
Please note: the suggestions below are just ideas for student experiments. I've trialled them all but I do not guarantee success. It is up to the teacher and student to see if they are practical for their school situation.
The syllabus allows for a broad range of experiments on acids and bases:
Science inquiry: investigate properties of acids and bases.
Students often suggest an experiment to compare the behaviour of monoprotic acids and polyprotic acids when being titrated against a strong base. This comes from the syllabus objective:
Properties of acids and bases • understand that acids are substances that can act as proton (hydrogen ion) donors and can be classified as monoprotic or polyprotic depending on the number of protons donated by each molecule of the acid. Reference: Chemistry 2019 v1.4, General Senior Syllabus, Queensland Curriculum & Assessment Authority, July 2022, page 40.
It also can be considered a modification (i.e. a refinement, extension or redirection) of the mandatory practical which states: "Acid-base titration to calculate the concentration of a solution with reference to a standard solution" (Syllabus, p 40).
Different acids have different numbers of protons (H+) that they can donate. This is a measure of the acid's 'protocity'. Monoprotic acids have a 'procitity' of 1. Ones commonly met in senior chemistry are hydrochloric acid (HCl) and acetic (ethanoic) acid (CH3COOH). For polyprotic acids, the most common are: (a) diprotic acids such as sulfuric acid H2SO4, and carbonic acid H2CO3. These are said to have a proticity of 2. (b) triprotic acids such as citric acid HOC(CO2H)(CH2CO2H)2 and phosphoric acid H3PO4. These have a proticity of 2.
The syllabus doesn't use the term 'proticity'. It just talks about 'the number of protons donated by each molecule of the acid'. It is not a mandatory term so you can choose, or not choose, to use it.
What are some possible research questions? The main question is how the titre of an acid related to its proticity. You could try to confirm the claim that a diprotic acid has twice the titre of a monoprotic acid when using the same titrant (eg 0.100 M NaOH), and the same sample size (eg 20.00 mL aliquot of the analyte), and the same temperature, and acid-base indicator (eg phenolphthalein) etc. You could extend this to ask if a triprotic acid has 3x the titre of a monoprotic acid.
What is the rationale behind titrations of monoprotic and polyprotic acids? This is vitally important. The syllabus says: "The distinction between strength and concentration should be covered" (page 40). Monoprotic acids such as the strong acid HCl and the weak acid acetic acid have one thing in common: they both have one hydrogen ion released when one molecule of the acid dissociates in water. That is, they have a protocity = 1.
A common misconception - be careful! Many students argue that the titre for a monoprotic weak acid will therefore be lower than for a monoprotic strong acid because there are not as many H+ ions in solution. This is completely wrong. It is true that there won't be as many H+ ions in solution before the titration begins. However, the titres will be exactly the same if we compare strong and weak acids of the same concentration (eg 0.100 M). Let me explain.
Even though a weak acid releases only some of its H+ ions when dissolved in water, as the titration proceeds these H+ ions are used up by the OH- ions from NaOH to form water. By Le Chatelier's Principle, the equilibrium shifts to the right, and more H+ ions are then released by the molecules. But these too are used up by the NaOH in the titration. By the end point all molecules have dissociated and all H+ have reacted. So, in a titration every molecule is made to dissociate by the end point. That is, 0.1 mol of acid will form 0.1 mol of H+ ions no matter whether it is a strong or weak acid. This also true for diprotic and triprotic acids: 1 mol of any diprotic acid (weak or strong) releases 2 mol of H+ ions in a titration. Likewise, 1 mol of any triprotic acid (weak or strong) releases 3 mol of H+ions in a titration. It doesn't matter whether they are strong or weak or somewhere in between. This is a common source of confusion for students.
How do we compare monoprotic and polyprotic acids experimentally?
The usual student thinking goes like this: If you made up standard 0.1 M solutions of a monoprotic acid (eg HCl), a diprotic acid (eg H2SO4) and a triprotic acid (citric acid), and titrated 20.0 mL aliquots of them with 0.100 M NaOH you should find the diprotic acid has a titre 2x that of the monoprotic acid, and the triprotic acid titre would be 3x the monoprotic acid. That may be good in theory but it may not make a successful student experiment. The reason is that the acids listed above are not primary standards so they can't just be weighed out and made up to a specified volume with water and their concentrations calculated. You can only do that with a primary standard.
A good primary standard needs to meet the following criteria:
Has a high level of purity Is very stable (won't break down) Has a high molar mass Is not likely to absorb moisture from the air Is non-toxic Is inexpensive and readily available
The second problem is about standardizing solutions of the acids. Even if you made up approximately 0.1 M solutions of the acids above, how would you standardize them to get the exact concentration? Most likely by titration you might say - but that is the very purpose of your experiment. You'd be going around in circles. It doesn't make sense.
However, many teachers will accept this limitation and just provide you with solutions of acids of the concentration you require. The lab technician may prepare and provide you with such solutions if you ask and if your teacher agrees. For example, you may request 0.100 M stock solutions of HCl, H2SO4, H3PO4 (phosphoric acid), citric acid, NaOH, and so on. If they are provided you would be wise to acknowledege the source of the acids provided and state that you assume that the concentrations are accurate. Otherwise the reader will wonder how you did it. Alternatively, you could check the accuracy of the provided solutions by titration against a primary alkaline standard such as sodium carbonate Na2CO3 (a diprotic base). All of this takes time, and you don't have much of that. Personally, I'd stick to two primary standards (KHP, and oxalic), and maybe, at a pinch, the triprotic citric acid - as I'll describe below.
The answer to the concentration conundrum is to find a monoprotic acid and a polyprotic acid that are primary standards and use them for your experiment. However, for senior chemistry, we have just two candidates that are commonly available in schools: potassium hydrogen phthalate (KHP) as the monoprotic acid, and oxalic acid dihydrate (COOH)2•2H2O as the diprotic acid. Both meet the criteria for primary standards shown above. Citric acid is not a primary standard but it is quite stable and has an assay of 100.0 ±0.5%, so we can say it is good enough as a primary standard for senior chemistry. The only impurities are water (0.02%) and sulfated ash (0.05%). Let's just use it - but only if you have time to include a triprotic acid in your design.
The 0.1M NaOH doesn’t have to be standardized but should be made up as accurately as possible (4.00 g/L) as you are just comparing the titres. Oxalic acid should have twice the titre of the KHP for equal aliquots (eg 20 mL) of the 0.1 M solutions. There is no triprotic acid I can think of that would be a primary standard. Phosphoric acid, while being triprotic, is not a primary standard. Oxalic acid is used as a rust cleaner and is quite hazardous. Make sure you speak to your teacher about whether it is safe for you to use and what controls you need.
The approach
1. Make up 500 mL of 0.1 M NaOH solution (4.00 g/L) and store in a sealed container to keep the CO2 out. You can use a measuring cylinder or a volumetric flask to measure out the water. Label correctly. 2. Make up 250 mL of 0.100 M KHP in a volumetric flask using deionized water, and also make up 250 mL of 0.100 M oxalic acid in another volumetric flask in the same way. Check whether you have the anhydrous or dihydrate form of oxalic acid. 3. Rinse glassware as appropriate (se hints below). 4. Using the pipette, take a 20 mL aliquot of the KHP and titrate using phenolphthalein indicator to a pink end point (pH 8.3-10.0). 5. Do replicate titrations until you have three titres within 0.10 mL (2 drops) of each other. These three titres are said to be 'concordant' titres. 6. Do the same with oxalic acid. 7. Do a 'blank' titration using 20 mL of deionized water instead of the acid. Include the phenolphthalein indicator.
8. Analyse your data as appropriate.
HINTS 0. Put safety glasses on. This is good practice anyway, but you will be filling a burette at eye level so best to be extra careful. 1. You could dry the KHP (on a watchglasses) in an oven at 100°C for 5 minutes and let cool (overnight if possible) in a dessicator over silica gel before weighing it out. You may not have a dessicator at school, or even silica gel, so this may be a problem. Let it cool in a sealed container if nothing else. If you don't have this equipment don't bother drying the chemicals. Do not try to dry the oxalic acid - it will decompose. If you're using citric acid you can dry it at 105°C as for KHP. Citric acid melts at 153°C and decomposes at 175°C so keep the temperature down. 2. Your school should have KHP and citric acid but may have to order in the oxalic acid, so give them plenty of notice. A risk assessment will tell you of its dangers and your school may not allow you to use oxalic acid. In that case - look for a different experiment. 3. Rinse out all glassware with deionised water at the start, including the tip of the burette. Bubbles wreck any experiment. 4. Then rinse out your burette with the NaOH solution twice. Open the stopcock to make sure the solution in the tip is also rinsed out. Place the burette in the clamp. Fill the burette just above the 0.0 mL mark. This should be done with the burette at eye level so you won't be looking up in the air. If you slip you could have NaOH in your face. Use a small plastic funnel if you like. Remove it during the titration though as it can trap droplets that randomly fall down inot the burette during the titration. Open the stopcock to fill the tip and ensure there are no bubbles of air in the tip. 5. Rinse out your pipette with the solution of acid. Let it drain for a short time before refilling. Some people like to do the acid rinsing twice. You only need to do this at the start of the series of KHP titrations. For the oxalic acid, rinse the pipette with water then the oxalic acid solution. 6. Fill the pipette to the mark (bottom of the meniscus) using a pipette filler (not your mouth). Transfer an aliquot of the KHP solution to the conical flask. Tip the flask so that the bottom of the flask is exposed and touch the tip of the pipette gently on the bottom. This will drain out a drop from the pipette and ensures an accurate amount of acid is in the flask. Do not tap the last drops out or shake them out. Put some white paper on the burette base to make the end point easy to observe.
7. Use a 20 mL pipette if you can. If all you have are 25 ml pipettes you may find the oxalic acid titration is more than 50 mL and that would mean refilling your burette. Besides being time consuming you would have additional sources of uncertainty (another ± 0.10 mL) as well as more chance of errors. If you have only 25 mL pipettes then I suggest you make your sodium hydroxide solution a little more concentrated from the start - perhaps 0.12 M. That would bring back your oxalic titration to 40 mL or so. 8. Add 3-5 drops of phenolphthalein indicator. What ever number of drops you choose, always use the same number for all titrations. Take an initial burette reading (IBR). Some students like to zero the reading by letting some NaOH out until the meniscus (or pointer) is at the zero mark. You don't have to. Any reading is okay so long as it is accurate.You should be able to read a 50 mL burette to the nearest half-scale division. That is, the scale divisions are 0.1 mL so you should be able to read to the nearest 0.05 mL. For example, if your initial burette reading is zero (0.00) you should record this with it's uncertainty of ± 0.05 mL. Write this down as IBR = 0.00 ± 0.05 mL. If you are not starting your IBR at 0.00 then you should have a second decimal place that is 0 or 5, such as 1.30 mL or 1.35 mL. 9. Titrate to a faint pink colour that lasts 30 seconds. Swirl as you go. See my video about this:
10. Take a final burette reading (FBR). As stated above, you should be able to read a 50 mL burette to the nearest half-scale division. Say you read the final reading as 21.35 mL you would record it with its uncertainty as 21.35 ± 0.05 mL. 11. Here are some examples of burette readings. Different brands of burettes may appear different and the general rule is 'read to the bottom of the meniscus (curve)'.
12. Watch for parallax errors. Your eyes should be at the same level as the mark on the burette you are reading and not above or below. Here are some examples:
13. To check that the titration is at the end point and you haven't gone past it, I always add one drop of the acid solution from the pipette to the flask. There will be one drop left in the tip of the pipette. It should change the solution back to colourless. If it doesn't, you have gone past the end point. Add another drop if this is the case and check again. If it takes 2 drops to go colourless then you are 1 drop (0.05 mL) past the end point. Subtract 0.05 mL from your titre.
14. Refill burette. Rinse the flask with deionized water. 15. Repeat steps above using exactly the same number of drops of indicator (3 drops) until you have three concordant titres. Concordant titres differ by no more than 0.1 mL from lowest to highest. 16. Concordant titres don't have to be one after the other. They could be the first and fifth titres. Check with your teacher about their definition of 'concordant'. The Oxford Chemistry for Queensland text (Unit 3 & 4, page 120) and most teachers will say 3 titres within a range of 0.10 mL, but they could expect something else. It might take you 5 titres to get this. Average the concordant titres and note the uncertainty. Here are 5 titres with the concordant ones (1, 4, 5) shaded in pink.
You wouldn't normally record all of the uncertainties but I've done it here to show you how the calculation works. The average of the concordant titres is (23.20 + 23.20 + 23.25)/3 = 23.22 mL. The uncertainty can be calculated by (max – min)/2 which equals (23.25 – 23.20)/2 = 0.03 mL. This less than the sum of two half-scale divisions (0.05 + 0.05 = 0.10 mL) and we always take the greater of the two. So we can report this average titre as 23.22 ± 0.10 mL. As percentage uncertainty this would be stated as 23.22 mL ± 0.4%. Note that percentage uncertainty is expressed to just one significant figure (one decimal place in this case). You might see why it is best to have titres around about the same volume as your aliquot (pipette volume). The larger the titre the lower the percentage uncertainty. In your report you could say why concordant titres are used rather than just averaging all titres. 17. Make sure you keep the NaOH in a bottle with a lid and try to do all titrations in the same double lesson. NaOH does go off as it absorbs CO2 from the air and forms sodium carbonate. This decreases the NaOH concentration somewhat. It also produces a buffer solution that interferes with the pH at the end point and the colour change is impacted. 18. In your report you need to say why phenolphthalein is suitable as an indicator (the end point has a pH in the 8.3 - 10 range and this is where phenolphthalein changes colour).
19. A 'blank' titration is done using a 20 mL aliquot of deionized water in the flask instead in the acid. This gives you a measure of how much the water itself contributes to the titre. You may have learnt that pure water has a neutral pH of 7.0 but in fact the deionized water in your lab is probably a bit lower than this as it absorbs CO2 from the atmosphere and forms the weak acid carbonic acid. This can make your deionized water have a pH as low as 5.5. However, it is likely to be in the 6 range. Phenolphthalein indicator changes colour to pink at a pH of about 8.3 to 9.0 so you always need a drop or more of base to get the water to that range. Ideally, 1 drop (0.05 mL) of the NaOH in the burette will change the colour to a faint pink that persists for 30 seconds, but it may require 2 drops (0.10 mL) or more. Just test it once and record the value. Ensure the flask is washed out well with water beforehand. This is a systematic error and should be deducted from your titre. Note that the blank titre will also have it a scale reading uncertainty of 0.10 mL, so you would record your titre (if it took 2 drops) as: V(blank) = 0.10 ±0.10 mL. Actually, when I do blanks I just count the number of drops of base I add from the burette and multiply that by 0.05 mL. I then just say my uncertainty is ±0.05 mL rather than ±0.10 mL.
For example, say I took a reading from the burette graduations: IBR 0.00 ±0.05 mL, FBR 0.15 ±0.05 mL, titre = 0.15 ±0.10 mL But if I count the drops: 3 drops = 3 x 0.05 mL = 0.15 ±0.05 mL. In the grand scheme of things it is not much different so either way is okay.
20. You probably won't get an average titre for oxalic acid that is exactly twice that of KHP but it should be close. For example if your KHP titre is 21 mL then your oxalic titre should be 42 mL or thereabouts. We expect a ratio of 2:1 for oxalic:KHP titres. If it is way different then you probably have made a mistake in preparing the solutions or doing the titrations. But that will allow you to explore why this might be and do a great discussion on uncertainty and errors.
21. If you want to take it further, you could monitor the pH of the titration by using a pH probe in the flask. However, you don’t need to do this to get high marks. For a strong acid vs strong base titration, the equivalence point is at a pH of 7.0 but the end point (when the phenolphthalein indicator changes to pink) will be a drop or two more as the indicator doesn't change colour until pH 8.3 or so. For a weak acid vs strong base the equivalence point is about pH 9.0 and the end point will be the same.
22. Lastly, if you are determined to do a triprotic acid then citric acid is the way to go. Your school probably has citric acid in the anhydrous form but you need to check the label.
The formula will be on the bottle and will state the amount of water of crystallisation (eg monohydrate, or anhydrous) and its molar mass. It will be about 99.5% citric acid with the rest just adsorbed water or contaminates from reaction with the air. If your school doesn't have any you can get it from the supermarket ($3 for 75 g) but it will probably not say on the label whether it is anhydrous or the monohydrate. If you put it in the oven at 50°C for an hour you will drive off the water of crystallisation (if there is any) and you can then be sure you have the anhydrous form.
Your titres should show that it is 3x the KHP titre, with an error of less than 1%. My results for citric acid are below.
EXPERIMENTAL RESULTS
We need to find out if these results support the claim that the titre for the diprotic acid (oxalic) is twice the titre for the monoprotic acid (KHP). In other words, is V(oxalic) = 2.00 x V(KHP). In doing so we need to find the uncertainty in this ratio. What we are really considering is whether the diprotic acid releases twice the amount of hydrogen ions than an equal volume of a monotprotic acid of the same concentration. Thus, the research question becomes: is n(H+) oxalic = 2.00 x n(H+) KHP?
1. KHP TITRATIONS (my results)
Average titre (KHP) = (18.85 + 18.95 + 18.90)/3 = 18.85 ± 0.10 mL. Concordant titres are in pink.
Note: to determine the uncertainty you can also use the formula for the concordant titres:
- which gives: (18.95 – 18.85)/2 = 0.05 mL. However, because the titres have to be concordant the maximum uncertainty will always be 0.10 mL. This is also the uncertainty in any single titre (i.e. a two lots of the half-scale division), so we just use ± 0.10 as the uncertainty in all titrations. If you had three titres exactly the same you would not use 0.00 as your uncertainty. You would use the larger value of ± 0.10 mL that applies to individual titres. It is better to overestimate the uncertainty than to underestimate it. The symbol for absolute uncertainty is the lower case Greek 'delta' δ, and the symbol for percentage uncertainty is δ%.
Blank titre = 0.10 ± 0.10 mL
Nett titre for KHP: V(KHP) = (18.85 ± 0.10 mL) – (0.10 ± 0.10) mL = 18.75 ± 0.20 mL (18.75 mL ± 1.1 %).
Notes: when adding or subrtacting values you always add the uncertainty. Also, uncertainty should be expressed to 1 significant figure (e.g. 0.5%, not 0.53%) unless the first figure is a 1 or a 2 and then you would use two significant figures, SF, (eg if the percentage uncertainty was 0.13% you would express it as 0.13%, but if it was 0.33% you would round it to 1 SF or 0.3%).
2. OXALIC ACID TITRATIONS (my results)
Average titre = 38.05 ± 0.10 mL. Concordant titres are in pink. Remember, the uncertainty of a titre is ±0.10 mL.
Nett titre for oxalic acid: V(oxalic) = (38.05 ± 0.10) mL – (0.10 ± 0.10 mL) = 37.95 ± 0.20 mL (37.95 mL ± 0.5 %).
3. RATIO OF HYDROGEN IONS IN THE KHP AND OXALIC SOLUTIONS BEING TITRATED
We can now determine the ratio of the amounts of hydrogen ions produced by each acid. To do this we use the titration results. We know that in a titration 1 mole of hydroxide ions (OH-) reacts with 1 mole of H+ ions. So we just calculate the number of moles of NaOH used in each titration. We could just use the titre volumes in the ratio because the concentration of the sodium hydroxide cancels out, but we should do this properly.
Moles of H+ ions in KHP titration: n(H+in KHP) = CNaOH(burette) x VNaOH(KHP titre)
Moles of H+ ions in oxalic acid titration: n(H+oxalic) = CNaOH(burette) x VNaOH(oxalic titre)
Conclusion: This confirms that a diprotic acid releases between 1.99 and 2.05 times the number of hydrogen ions compared to a monoprotic acid.
We could just leave out experiment there and report that result, but it would be better to look at the uncertainty associated with the solutions of acids prepared in this experiment and not just the uncertainty in the titres.
What about the triprotic acid citric acid? As I said, citric is not a primary standard but it is close to 100% so should be okay. It may have adsorbed water or may have some breakdown products included. However, the manufacturers say (on the bottle I have) that it is at least 99.5% pure. So I have included my titres just in case you are tempted to include citric acid. Net titre (after subtracting the blank: 56.25 ± 0.20 mL (56.25 mL ± 0.4%). I had to refill my burette so there were four lots of scale reading uncertainty (4 x 0.05 = 0.20 mL).
EXPECTED RESULTS
We know that theoretically a diprotic acid releases two times the number of hydrogen ions compared to a monoprotic acid but what is a reasonable value for an experimental test of this ratio? In this section we will calculate the uncertainty in the concentration of the original solutions and the amounts transferred to the flasks for the titration.
1. KHP solution Mass of KHP: m(KHP) = 5.10 ± 0.01 g = 5.10 g ± 0.2% Molar mass of KHP: Mr(KHP) = 204.2 g/mol Volumetric flask: V(flask) = 250.0 ± 0.10 mL (manufacturer's specification) = 0.250 L ± 0.04% Pipette: V(pipette) = 20.0 ± 0.02 mL (manufacturer's specification) = 0.020 L ± 0.10%
We know that 1 mole of KHP theoretically releases 1 mole of hydrogen ions:
2. Oxalic solution Mass of oxalic acid dihydrate: m(oxalic) = 5.10 ± 0.01 g = 5.10 g ± 0.2% Mr(oxalic) = 126.07 g/mol Volumetric flask: V(flask) = 250.0 ± 0.10 mL (manufacturer's specification) = 0.250 L ± 0.04% Pipette: V(pipette) = 20.0 ± 0.02 mL = 0.020 L ± 0.10%
We know that each mole of oxalic acid theoretically releases 2 moles of hydrogen ions: 3. Oxalic acid to KHP ratio We can now calculate the theoretical mole ratio of hydrogen ions in the 20 mL aliquot of oxalic acid compared to the hydrogen ions in the 20 mL aliquot of KHP:
Conclusion: The accepted (theoretical) range for the ratio of hydrogen ion released by a diprotic acid to that of a monoprotic acid in an experimental test is 1.98 to 2.02. We found that experimentally the range was 1.99 to 2.05 which overlaps the accepted values. We can confidentally say that we have confirmed the claim that diprotic acids release twice the amount of hydrogen ions as monoptotic acids as shown by the average titre for oxalic being twice the average titre of the KHP.
ACCURACY Lastly, we should express the absolute and percentage error of our results. That is, the difference between the theoretical value (2.00) and our experimental value (2.02). The calculations are as follows:
What can I graph to display my results? You don't always have to include a graph in your report. The syllabus criteria for the highest level talks about 'visual and graphical representations of data' that shows 'correct and relevant processing of data', and 'thorough identification of relevant trends, patterns or relationships' (p 54). So graphs are not essential to get the highest marks, but if they help you demonstrate trends or relationships then they could be warranted. In our case, a graph of protacity (x-axis) as the independent variable, and titre (y-axis) as the dependent variable would be appropriate. The proticity scale should be in whole numbers (0, 1, 2, 3) with not fractions in between as you can't get a procity of 1.5 for instance. It is a 'discrete' variable, unlike titre which is a 'continuous' variable. Here's my graph, including citric acid:
Why doesn't the graph's trendline go through zero? The graph shows that the titre is directly proportional to the proticity, as expected. The gradient of 18.75 tells us that the titre volume increases by 18.75 mL for each additional value of proticity. What is especially interesting is the y-intercept (c-value) of 0.15 mL. Normally, this represents the systematic error in the investigation. For example, a solution with a proticity = 0, such as water, should have a titre of 0.00 mL. But you should recall that when we did the blank titration with 20.00 mL of water the phenolphthalein indicator didn't change colour until one or two drops of the titrant (0.100 M NaOH) was added. This is called the 'blank' and is subtracted from the titre. This is the volume of 0.100 M NaOH (the 'titrate') in mL that takes the 20.00 mL of water (pH 6-7) to the 'end point' (pH 8.3 - 10.0) where the indicator changes colour.
The y-intercept value of 0.15 mL is saying that the line of best fit is precicting a value of 0.15 mL (3 drops) for a proticity of zero. This could be an error in our technique that says our blank should have been 3 drops more of NaOH. I found it was 3 drops but maybe it was more like 6 drops. It could also be an error in the assumption about the citric acid that it is a primary standard. We know it is not a primary standard and if you read the data sheet for it we are told that it is 99.5% pure. This means that what we called 0.100 M citric acid is really only 0.0995 M. Thus, the titre is for 20.00 mL of 0.0995 M citric acid wheras the other titres are for 0.100 M KHP and 0.100 M oxalic acid. So the real titre for citric acid should be a little bit greater than 56.25 mL (maybe 56.40 mL). That gives a trendline that goes through zero and increases the R2 value. Problem solved.
How can the experiment be improved? By the time you get to this stage of your report writing you are probably heartily sick of it all. All students are. But, for improvements don't just throw in all sorts of random ideas. When you suggest an improvement make sure they are relevant to your experiment and that they will be effective (that is, they should make a difference). Common ones are 'do more trials', or 'use a more accurate burette' but will these make a difference? In essence, what you are trying to do is to reduce the uncertainty of the results, providing your method is valid. Doing more trials reduces random error but it adds a lot more time. Students say 'use a 25 mL pipette rather than a 20 mL one'. This helps because a bigger burette means a bigger aliquot and a bigger titre. Increased sample size always helps. These decrease the percentage uncertainty of the measurement.
2. CONDUCTIVITY EXPERIMENTS
One of the interesting properties of acids and bases is their electrical conductivity. That is, how well do solutions of acids and bases conduct electricity. To keep it simple, we can just consider acids. How does their conductivity depend on whether they are weak or strong, dilute or concentrated.
These questions can provide a wide range of research questions for a student experiment that fits within the Unit 3 syllabus objectives:
Properties of acids and bases • Discriminate between the terms strong, weak, concentrated and dilute for acids and bases. • Discriminate between strong and weak acids and bases in terms of the extent of dissociation, rate of reaction, pH and electrical conductivity. Reference: Chemistry 2024 v1.0, General Senior Syllabus, Queensland Curriculum & Assessment Authority, Januiary 2025, page 33.
Do polyprotic acids have higher electrical conductivity than monoprotic acids? You would think so because polyprotic acids, by definition, can release more hydrogen ions than monoprotic acids. In this section we will compare the degree of ionization of the acids by measuring their electrical conductivity. Recall that not all of the molecules of the acid ionize in solution. Strong acids do, but weak acids are only partly ionized.
What is electrical conductivity? It is a measure of the ease at which an electric charge can pass through a solid, liquid or gas. Copper wires have high electrical conductivity; salty water has medium conductivity; and plastic - has none.
What are some research questions we could ask about acids and conductivity? Here are some that students usually ask, and I've put a quick response in brackets to guide you in developing a research question that you have time and equipment for. In all cases I'm comparing equal concentrations of the dilute acids, eg 0.100 M.
1. Does a strong diprotic acid (H2SO4) have twice the conductivity of a strong monoptotic acid (HCl, HNO3)? (Answer: no) 2. Do all strong monoprotic acids (HCl, HNO3) have the same conductivity? (Answer: no) 3. Does a weak diprotic acid (oxalic acid) have twice the conductivity of a weak monoprotic acid (acetic acid, KHP)? (Answer: no) 4. Do all weak monoprotic acids (acetic acid, KHP) have the same conductivity? (Answer: no) 5. Could a weak triprotic acid (citric acid, phosphoric acid) have greater conductivity than a strong monoprotic acid (Answer: could do). 6. Does the conductivity of weak acids increase as you go from monoprotic, to diprotic, to triprotic? (Answer: could do) 7. How does conductivity of a weak acid change with temperature? (Answer: could go either way - see note at end).
In the suggestions above, I've detailed experiments on the relationship between titres of acids and their proticity. No regard was given for whether the acids were weak or strong, as that aspect makes no difference in a titration.
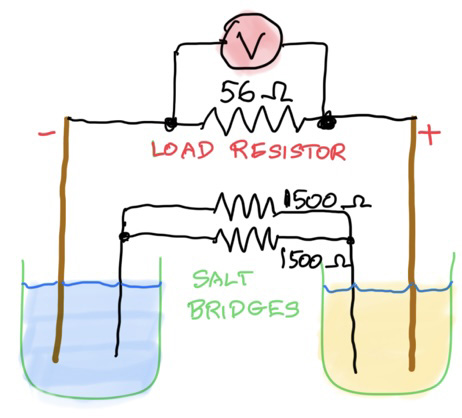
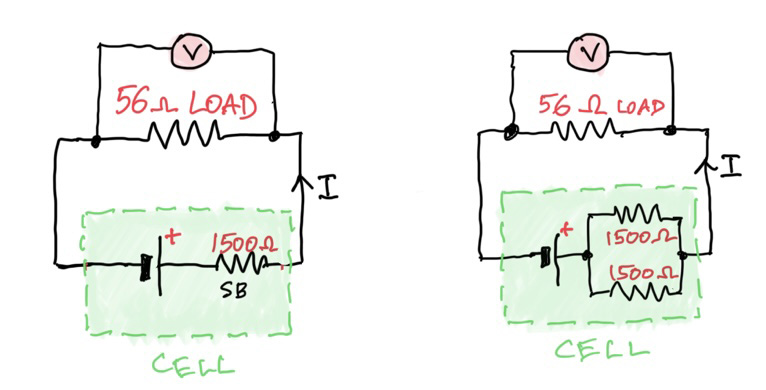
How do you measure electrical conductivity of a solution of an acid? Conductivity is measured using a conductivity device in which an electrical current is passed through a solution and its electrical current measured. The greater the current for a given voltage the greater the conductivity. You can use a conductivity probe, or set one up yourself with the help of some simple equipment. The diagram below is the basis of all conductivity measurements:
By placing two inert electrodes a small distance apart in the acid, and applying an alternating voltage across them, the amount of current that passes through the solution is a measure of its electrical conductivity (κ, kappa, in the units of siemens (S) per centimetre, S cm-1, or more commonly microsiemens per centimetre, µS cm-1). In some books they use the Greek letter sigma, σ, for conductivity.
Students ask "do we need to use alternating curent; will DC be okay?". I'll say now: "AC is essential if you want sensible results" and will explain why in the experimental design section later.
What is the conductivity formula? To determine the conductivity of a solution you need to know the voltage (V, in volts), the total surface area of the electrodes (A, in cm2), the current (I, in ampere, A) and the distance apart (L or l, in cm). I've used capital L in these notes as the lowercase version 'l' can be confusing. Note that the units are not all SI. For convenience and for historical reasons the length unit has been kept as centimetres (cm). The formula is:
If you then divide this value by the concentration in mol/L it is then called the molar conductivity. Note: If you look up the definition of conductivity you should type in 'electrical conductivity' otherwise you may get 'thermal conductivity' which is a measure of thermal (heat) energy flow in a substance.
Do we need to work out conductivity for our student experiment (IA2) or will the current be okay? You can work in terms of the current (in ampere, A). That is perfectly fine and makes for a less complicated report. I'm calculatimng conductivity but that may be hard for you to do the calculations. I'll explain later.
What are some accepted values for conductivity? Conductivity is usually expressed in microsiemens per centimetre (µS cm-1). It is often called 'specific conductivity' but the term 'conductivity' is always understood to mean 'specific conductivity'. Here are the accepted values of some common acids, all at a concentration of 0.100 M. Source: CRC Handbook of Chemistry and Physics, 97th edition, 2017, section 5-74. This book is not open access but copies can be found on the internet.
Specific conductivity, σ (µS cm-1)
Because they are all 0.100 M, the molar conductivity would be the specific conductivity value (above) divided by 0.100, e.g. HCl would have a molar conductivity of 355280 (µS cm-1 M-1 or µS cm2 mol-1). Molar conductivity has the Greek uppercase symbol lambda, Λ, so the formula becomes: Λ = κ/C. Whew!
What does electrical conductivity measure for ionic solutions such as acids? It is a relative measure of the concentration of ions in the solution. It is not necessarily directly proportional but increases with an increasing number of ions. The size of the ions can affect the motion of the charge, with big ions making for lower conductivity. The amount of hydration (number of attached water molecules) also affects the size of an ion.
EXPERIMENTAL DESIGN Using a conductivity sensor. Common ones used in schools are the Pasco wireless conductivity sensor, and the Vernier Conductivity Probe. They are great but cost about $350 and not all schools can afford that, especially if you have six groups in your class.
Using a voltmater and ammeter. If your school doesn't have a conductivity sensor or meter, it is quite likely they will a simple conductivity apparatus available. Here are two I have used in the past:
Making your own conductivity apparatus As I said, there are many types available but not all schools will have them. Here are some electrode types:
Platinum electrodes are ideal for conductivity experiments as they are very non-reactive. However, platinum is very expensive and your school won't have any. You can buy platinized titanium mesh electrodes from Ali Express for a few dollars each if you are keen. They are used in gold or rhodium plating of jewellery. Its too complicated so let's move on.
Carbon rods such as those found inside a 1.5 V carbon-zinc dry cell would be okay, however, it's impossible to buy the D-size carbon-zinc batteries these days as they are invariable alkaline batteries and have no carbon rod. Your school may already have carbon rods supplied by one of the science companies (eg Haines $3.40 each, but minimum spend of $30). To get graphite rods, I bought a 6 mm diameter graphite rod (25 cm long) from an art supply company for $3.50. Cut it in half and that works well (see below). The problem with graphite rods is that they are round so it is difficult to work out the surface area and distance apart. Nevertheless, I made one up for the purpose of showing you how they work. One problem is that you need to keep the electrodes in the solution to the same depth for all tests. This is because the surface area of electrodes is a variable and you want to control it. It is hard to keep the depth exactly the same for all solutions. At school, I tell students to measure in 2.0 cm from the end and wrap some masking tape around the electrodes. That way only 2 cm is exposed to the solution. After many years I found it best to use heat shrink tubing to cover most of the rod, then the rods could be used year after year. Heat shrink tubing (6 mm diameter) is available from Jaycar for $2.45.
My setup is pretty simple and one I have used in class time and time again. I like it for student experiments because you don't need to worry about corrosion from the surface affecting the results. Graphite (carbon) is not reactive.
So, to work out the conductivity I use the formula:
Stainless steel makes a good electrode as it is fairly non-reactive and, best of all, you have control over surface area and separation distance. Stainless steel rulers are made from the '304' alloy which has a high nickel content (18%) and thus highly resistant to corrosion, especially in electrolysis cells. If you can get some plain stainless steel plate then well and good. However, I bought a 304 stainless steel ruler for 99 cents at Officeworks and cut two 10 cm long pieces off it and ground the painted letters off the ends with a wire brush in a drill. I covered a 2 cm long section of each end with masking tape and sprayed it all with several coats of paint and then removed the tape. I placed a small piece of wood between the electrodes and held them together with a rubber band.
I connected the electrodes to the two meters and the power supply as shown in the diagram above. I made up 50 mL solutions of the acids and had them in specimen jars. Then I positioned the electrodes so the exposed areas were under the surface of the liquid.
Worked example Calculate the conductivity in µS cm-1 (microsiemens per centimetre) of a 0.100 M solution of KNO3 in a conductivity apparatus using two plates each measuring 2.5 cm x 2.00 cm with a separation distance of 1.26 cm. A potential of 10.01 V AC is placed across the electrodes and gives a current of 0.853 A.
HINTS: 1. Use AC electricity, not DC if possible. Direct current (DC) will work and will give you usable results and a linear trendline. You will see expected trends between solutions but your values won't be close to the accepted values. Here are the main reasons:
(a) Water will hydrolyse (breakdown). If you have to use a DC power supply you would have to keep the voltage below 1.23 V. This is the standard reduction potential for water so any voltage greater than this will cause the water to hydrolyse (by electrolysis) into hydrogen gas and oxygen gas. Actually, to hydrolyse water, you need a voltage a little higher than this as there is 'overpotential' which means some energy is lost in the electrodes and the electrode-water interface. The higher the voltage the faster the decomposition occurs. Here's a photo showing the bubbles forming at just 2.0 V DC. You really want to avoid this. It will affect your experiment. The result of this is that the concentration of ions increases with time. So you should get a reading the instant it is turned on and not let it sit there hydrolysing for too long. Hydrolysis causes bubbles of oxygen gas to form on the anode at a potential over 1.23 V. Here they are shown at 2.0 V DC for 0.100 M citric acid. There were similar bubbles of hydrogen on the cathode. These microbubbles prevent the solution from coming in contact with the electrodes and lowers the conductivity. Comparison of DC and AC electricity used in measuring the conductivity of 0.100 M citric acid. Note that for AC at 9.0 V, the current stays at 173 mA for the duration. However, with DC at 9.0 V, the current rises as more ions are prodiced. This means the conductivity is changing as you try to measure it. It is not a big change but one worth noting. As well, the temperature changed from 25°C to 29°C for DC but only to 28 °C for AC. (b) Low current is hard to measure. The problem with DC is that with a voltage less than 1.23 V, the current is very low. Pure water, and even electrolyte solutions will not conduct under 1.23 V and if they do it must be unknown reactions taking place. It will at best a current of about 15 µA. That's 0.000015 A. That's only two significant figures and an uncertainty of ± 1µA. That's a scale reading uncertainty of 7% so you're off to a bad start already. (c) Change in pH. With hydrolysis of water H+(aq) and OH- (aq) ions are produced at the cathode and anode respectively. There is an increased pH next to the anode due to the production of OH- ions. These ions lessen the production of oxygen gas and slows the reaction down. This reduces the current. I added phenolphthalein to each conductivity cell to show that AC doesn't produce more OH- ions than H+ ions as the indicator stayed colourless. However, DC turned the indicator pink. This may seem strange as H+ and OH- ions are supposed to be produced in equal amounts (so no colour change). I think some of the Fe in the steel ruler was oxidized to Fe2+ at the cathode instead of producing H+. Thus there is more OH- produced in the cell than H+ so it turns pink. (d) Polarisation. Direct current depletes the ions in the electrolyte solution near the electrodes. This is called polarisation. The lack of ions means higher resistance and thus lower conductivity than it really is. (e) Hydrated ions and their big hydration shell. Water molecules attach themselves to the ions in solution and we say they are hydrated ions. For example, H+(aq) ions are usually represented a H3O+ to show that the ion is hydrated (attached to a water molecule). Actually, scientists aren't sure how many water molecules are attached to each hydrogen ion - it could be two (H5O2+) or more. When ions are made to move by the electric field between the electrodes, the ions have to drag this big 'hydration shell' along with them as they migrate from one electrode to the other. Its like running with a backpack on. Of course you're going to go slower. So the current is reduced. The other problem is that the hydration shell can change geometry during the process. It could go from a octagonal shell to a hexagonal shell and that process is endothermic. It is just one big uncontrolled hassle. (f) Electrolysis of other ions. With some ionic solutions their breakdown (electrolysis) voltage can be quite low, so you have to be even more careful. For example, if you were measuring the conductivity of a CuSO4 solution, you'd have to keep the voltage below 0.89 V.
(a) Water will hydrolyse (breakdown). If you have to use a DC power supply you would have to keep the voltage below 1.23 V. This is the standard reduction potential for water so any voltage greater than this will cause the water to hydrolyse (by electrolysis) into hydrogen gas and oxygen gas. Actually, to hydrolyse water, you need a voltage a little higher than this as there is 'overpotential' which means some energy is lost in the electrodes and the electrode-water interface. The higher the voltage the faster the decomposition occurs. Here's a photo showing the bubbles forming at just 2.0 V DC. You really want to avoid this. It will affect your experiment. The result of this is that the concentration of ions increases with time. So you should get a reading the instant it is turned on and not let it sit there hydrolysing for too long.
(b) Low current is hard to measure. The problem with DC is that with a voltage less than 1.23 V, the current is very low. Pure water, and even electrolyte solutions will not conduct under 1.23 V and if they do it must be unknown reactions taking place. It will at best a current of about 15 µA. That's 0.000015 A. That's only two significant figures and an uncertainty of ± 1µA. That's a scale reading uncertainty of 7% so you're off to a bad start already.
(c) Change in pH. With hydrolysis of water H+(aq) and OH- (aq) ions are produced at the cathode and anode respectively. There is an increased pH next to the anode due to the production of OH- ions. These ions lessen the production of oxygen gas and slows the reaction down. This reduces the current.
(d) Polarisation. Direct current depletes the ions in the electrolyte solution near the electrodes. This is called polarisation. The lack of ions means higher resistance and thus lower conductivity than it really is.
(e) Hydrated ions and their big hydration shell. Water molecules attach themselves to the ions in solution and we say they are hydrated ions. For example, H+(aq) ions are usually represented a H3O+ to show that the ion is hydrated (attached to a water molecule). Actually, scientists aren't sure how many water molecules are attached to each hydrogen ion - it could be two (H5O2+) or more. When ions are made to move by the electric field between the electrodes, the ions have to drag this big 'hydration shell' along with them as they migrate from one electrode to the other. Its like running with a backpack on. Of course you're going to go slower. So the current is reduced. The other problem is that the hydration shell can change geometry during the process. It could go from a octagonal shell to a hexagonal shell and that process is endothermic. It is just one big uncontrolled hassle.
(f) Electrolysis of other ions. With some ionic solutions their breakdown (electrolysis) voltage can be quite low, so you have to be even more careful. For example, if you were measuring the conductivity of a CuSO4 solution, you'd have to keep the voltage below 0.89 V.
2. Use alternating current (AC) if at all possible. Alternating current causes the anode and cathode to reverse polarity 50 times a second (if you are using a normal laboratory AC power supply). That's a frequency of 50 hertz (50 Hz). Commercial units have a default frequency of 25 kHz. Bubbles never get a chance to form as the electrode is constantly changing its mind. Also, you don't have to worry about the effect of ionic size and how this will hamper conductivity. So we get a more stable reading if this phenomena is eliminated as it is with AC. There is a small 'capacitance' effect with AC whereby some energy is lost as the ions jiggle around - but it is not a problem for senior chemistry.
3. Current is non-linear with voltage. Electrolyte solutions such as weak acids are said to be non-ohmic. When you test the conductivity of a solution with a low voltage (< 2V) it appears that the conductivity is very low, but at higher voltages the conductivity appears to be higher. This means the ratio I/V is not constant. The voltage you choose is very important. The higher the voltage the less the error so I suggest you use about 5-10 V. For example, I tested a 0.100 M solution of KNO3 which has an accepted conductivity at 25°C of 11120 µS cm-1 and got a conductivity of 4500 µS cm-1 at 1.0 V (60% error) but a value of 10700 µS cm-1 at 5V (4 % error). So, the higher the voltage the more accurate the readings. However, at high voltages the current is usually quite high (maybe 1 A) and if the wires start smoking, turn it down to 5 V or so.
4. Use a voltage divider. Common laboratory power supplies have a switch to select voltages in 2 V increments from 0-12 V but they are only approximate, and only give the selected voltage when nothing is connected. This is called a condition of 'no load', or open circuit. For example, the IEC power supply common in schools when set on 8V gives an actual voltage of 8.7 V AC, and 9.5 V DC. If you want to keep the voltage constant (very important for a fair test), eg 10.00 V, you need a device that allows you to make fine adjustments. Its called a potentiometer. Details are below. There are many types of potentiometers available. Your school should have several but are probably the big ones - and these work well.
5. Decide on a voltage and keep it constant. This is crucial if you want a fair test and to get results that make sense. Say, for instance, you find that 5 V AC (or DC) is a good voltage for your experiment, then you want to test every solution with the voltmeter (across the conductivity electrodes) reading 5.0 V every time. You can adjust the voltage to 5.00 V before you place them in a solution using a potentiometer as I have mentioned above. The ammeter will read 0 A of course. However, when you dip the electrodes into your solution current will start to flow and the voltage reading will drop because it is now under load. You need to bring the voltage back up to 5.00 V using the potentiometer and then take your current reading off the ammeter. It must be at your chosen voltage when the electrodes are in solution. It will drop to different values depending on the conductivity of the solution, so you can't just 'set and forget'. For example, when I tested 0.100 M KNO3 solution, the voltmeter dropped from 5.00 V to 4.37 V. I then had to wind it back up to 5.00 V and take a reading on the ammeter (0.340 A). However, using the more concentrated 1.0 M KNO3 solution, the voltage dropped from 5.00 to 2.95 V because it was more conductive and allowed a bigger current so it put a bigger load on the power supply. When I wound it back up to 5.00 V AC I could take my ammeter reading (1.79 A).
The reason for this is due to the internal resistance of components in the power supply. They have an internal resistance of 2 ohm (2 Ω). When current flows through the external circuit, some voltage is lost in this internal resistance. The bigger the current, the bigger the voltage loss. For example, when the current is 0.1 A, the voltage loss by Ohm's Law (V = I x R) is 0.1 x 2 = 0.2 V so the meter drops from 5.0 V to 4.8 V and has to be adjusted back to 5.0 V. When the current is 1 A, the voltage loss is 1.0 x 2 = 2 V so the meter drops from 5 V to 3 V, and has to be wound back up to 5 V. I've seen lots of student reports where they have just set the power supply to a nominal 6 V and never checked the voltage again.
If your experiment involves strong acids like hydrochloric acid or sulfuric acid you may find your power supply can't deliver a voltage as high as 10 V because of the high current and voltage drop. A lower voltage may be necessary, and then you'd have to use that for all samples.
6. Do I need to work out conductivity - isn't reporting curent enough? Yes, reporting just current is fine so long as the voltage is constant (which, for AC, it probably won't be). It is best to record both the voltage AND current for each trial and then calculate the conductance C from that. Conductance is just the current divided by the voltage (C = I/V). It is the reciprocal of resistance which is R = V/I). For example, if the voltage was 4.95 V and the current was 1.20 A, then the conductance would be C = I/V = 1.20/4.95 = 0.24 S. The unit symbol 'S' stands for 'siemens' which is the reciprocal of resistance (C = 1/R). To work out conductivity κ (as distinct from conductance) you need to know the surface area of the electrodes and the distance apart. Sometimes it is hard to work this out, especially if your school's conductivity apparatus consists of round electrodes. In this case you could just compare the conductance, C, for different acids.
7. Work out a 'cell constant'. I wouldn'ty bother doing this as it gets too complicated - but here's a different approach if you want toResearchers often don't bother with measuring the area and distance apart of the electrodes. They determine the 'cell constant' instead. It has the symbol G*. This is the value of L/A for the cell. You can do this by using a standard 0.100 M solution of potassium chloride (KCl) at 25°C in your experiment. KCl is a primary standard so an accurate concentration can be made up in the lab. It has a conductivity of 11121 µS cm-1 at 25°C, so if you measure the voltage and current you can work out the cell constant. Then use this in the calculations of conductivity (σ) for your acid solutions. The equation would be: σ = (I/V) x G*. For example, you could do as I did and use a 0.100 M KNO3 solution which has an accepted conductivity of 10700 µS cm-1 at 25°C. I got a current reading with my setup of 0.340 A at 5.00 V. This gave me a cell constant G* of 10700/(I/V) = 10700/(0.340/5.00) = 157353 cm-1. So, for any other readings I just multiply my current (in A) by 157353 and then divide by the voltage (5.0 V). For example, when I measured the conductivity of 0.100 M KHP solution (a weak acid) at 25°C I used 5.00 V and got a current of 0.303 A. My conductivity is then 157353 x 0.303/5.00 = 9416 µS cm-1. The accepted value is 9803 µS cm-1, an error of just 3.9 %. Alternatively, you could keep it simple and just report the current. You can still get full marks.
Worked example. A conductivity cell is filled with 0.100 M KCl at 25°C. A current of 0.680 A passes when a voltage of 10.0 V is used. Calculate (a) the cell constant; (b) the conductivity of a dilute acetic acid solution (in µS cm -1) which gives a current of 63.8 mA when 10.0 V is impressed across the electrodes. Note: the accepted conductivity of 0.100 M KCl at 25°C is 11121 µS cm-1. Answer. (a) G* = σKCl x V/I = 10749 x 10.0/0.680 = 158277 µS cm-1 (158000 µS cm-1 to 3 SF). (b) σacetic = G* x I/V = 158277 x (63.8 x 10-3)/10.0 = 1009.8 µS cm-1 (1010 µS cm-1 to 3 SF).
Worked example. A conductivity cell is filled with 0.100 M KCl at 25°C. A current of 0.680 A passes when a voltage of 10.0 V is used. Calculate (a) the cell constant; (b) the conductivity of a dilute acetic acid solution (in µS cm -1) which gives a current of 63.8 mA when 10.0 V is impressed across the electrodes. Note: the accepted conductivity of 0.100 M KCl at 25°C is 11121 µS cm-1.
Answer. (a) G* = σKCl x V/I = 10749 x 10.0/0.680 = 158277 µS cm-1 (158000 µS cm-1 to 3 SF). (b) σacetic = G* x I/V = 158277 x (63.8 x 10-3)/10.0 = 1009.8 µS cm-1 (1010 µS cm-1 to 3 SF).
2. CONDUCTIVITY OF WEAK AND STRONG MONOPROTIC ACIDS
In the suggestions that follow I'm looking at comparing the conductivity of a strong monoprotic acid (eg HCl) with that of a weak monoprotic acid (eg acetic acid).
NOTE: as I said before, you do not necessarily have to calculate the numerical value for conductivity of the solutions tested. You could just keep everything (voltage, area, distance) constant and just compare the currents shown on the ammeter. You are trying to show the relative values of conductivities rather than their absolute values, so comparing currents would be quite appropriate. However, working out the absolute values is a bonus and may give you more to discuss.
What factors affect the ionization of acids in solution? Strong acids are those that almost fully ionize in solution. For polyprotic strong acids (H2SO4) we are only talking about the first ionization of the acid. For sulfuric acid (a strong diprotic acid) it is only the first ionization that is 100%: H2SO4 → HSO4-(aq) + H+(aq). There is a second ionization that is not complete (not 100%): HSO4-(aq) → SO42-(aq) + H+(aq). We can add them together to get the total amount of H+released.
For example, when 0.100 mol of the strong acid HCl is added to water and made up to 1.0 L, the covalently bonded H-Cl molecules break apart and we will have 0.100 mol of H+ released as well as 0.100 mol of Cl- released. The same goes for nitric acid - another strong acid. However, when 0.100 mol of sulfuric acid (a strong diprotic acid) is added to a litre of water, it will release 0.100 mol by the first ionization but only a smaller extra amount of about 0.029 mol is released by the second ionization. This gives a total of 0.129 mol in a litre of solution, or 0.129 M.
How do we calculate the [H+] in 0.100 M solutions of the weak acids? You need to use the acid dissociation constant Ka. We quantify the amount of weak acid molecules that dissociate by their acid dissociation constant, Ka (see Syllabus p 41). That doesn't mean you have to use it in your report although you would be expected to perform calculations in an external examination. The syllabus makes this clear:
Check with your teacher about the level of detail required in a student experiment report.
Nevertheless, consider the weak acid, acetic acid, CH3COOH. It will partially dissociate into the cation H+(aq) and the anion CH3COO-(aq). It is easier to represent as HA → H+(aq) + A-(aq). The amount of the three species in a solution of a particular concentration (eg 0.100 M) is given by the relationship:
This makes the solution to the equation much simpler as it is no longer a quadratic. Note that this approximation only works for weak acids. If you want to be exact, you should use the quadratic form x2 + Kax − KaC = 0. Let's make the assumption and continue:
Example: Calculate the total ionic concentration of a 0.100 M solution of acetic acid (Ka = 1.76 x 10-5). Answer: Using the approximation for when Ka is very small [Note: you should state this]:
2. CONDUCTIVITY OF MONOPROTIC AND POLYPROTIC ACIDS
Experimental results Here are my results. I used two graphite electrodes with an exposed surface area of 2.0 cm2 on each giving a total area, A, of 4.0 cm2, and had them a distance, L, 0.90 cm apart. Conductance C = I/V. Conductivity κ = conductance x 0.90/4.0. I used an AC power supply and set the voltage at 10.00 V but it drifted depending on the conductivity of the solution. I tried 5 V but the current was too small. The current, I, readings are shown in the table.
How do they my experimental results compare to the total ion concentration of the solutions? Here I've shown the total ionic concnetration of the different acids (at 0.100 M) and my experimental results.
What relationship is there between ionic concentration and experimental measures of conductivity? The graph below shows the relationship between my experimental results and the ion concentration for the acids.
How do my experimental conductivity results compare to the accepted value? I thought I'd compare my expeimental results with the accepted values. The closer the gradient is to 1.00 the better the accuracy between experimental and accepted.
Accepted Conductivity, κ (µS cm-1)
The graph has a gradient of 0.98 which shows that there is an percentage error of 2% between experimental results and accepted results. We are able to predict the accepted value with confidence just by subtracting 2% on to the experimental values. The high R2 value of 0.98 shows that there is little uncertainty in the results. Any value above 0.95 is considered precise enough for senior chemistry.
CONCLUSION You would not be wise to do an experiment as time consuming as this if it is for a student experiment in senior chemistry. I've done it because I wanted to show you that you can test the relationship between ONE pair of acids as mentioned in the seven questions earlier. Just pick one pair of acids and compare the experimental conductivities, or even just the ratio of currents, and maybe look at the theoretical amounts of dissociation. You could compare your conductivity results to the accepted values to work out percentage errors.
When canning vegetables such as beetroot, it is essential to get the pH right to prevent the growth of bacteria. Beetroot is canned in a vinegar and sugar syrup and the cannery needs to get the pH between 3.9 and 4.0, and the titratable acidity of 0.9% acetic acid (0.15 M). For example, Golden Circle cannery in Brisbane makes a syrup of 14000 L water, 2.5 L 30% acetic acid, 1.7 tonnes of sugar (2 cu m). Final volume is 15000 L, [H+] 1.26 x 10-4 M, pH 3.9, sugar 12%.
However, adjusting the pH of a weak acid solution is not easy. How does the equilibrium shift as it is diluted? The QCAA 2025 chemistry syllabus says:
One way we can see how equilibrium shifts when a weak acid is diluted is to measure it conductivity. This makes a great prac.
How does electrical conductivity vary with the concentration of a weak acid? We know that a solution will conduct electricity because of the presence of ions in solution. For acetic acid - a weak acid - the ions are H+(aq) and CH3COO−(aq). However, acetic acid doesn't fully dissociate when dissolved in water. The percentage of CH3COOH molecules that ionize depends on the concentration of solution and the relationship is shown by the acid equilibrium constant Ka.
One good measure of ions present in a solution is the electrical conductivity. A high concentrations of ions means high conductivity, and vice versa. A good experiment is to measure the conductivity of a series of solutions of different concentrations of a weak acid such as acetic acid. Acetic acid is said to be monoprotic which means that each molecule of CH3COOH releases one C ion when it dissociates. It also releases one anion as well so the reaction is: CH3COOH → CH3COO− + H+. So, one molecule of CH3COOH produces two ions, or 1 mole acid produces 2 moles of ions.
EXPERIMENT. Firstly, I made up a 100 mL of 1.00 M solution of acetic acid. You would be provided with this if you asked for it. I then standardized it against 0.100 M NaOH using a phenolphthalein indicator. I made up eight accurate dilutions as shown in the table below. I found it easier to weigh out the amounts needed as the density is the same as water at this dilution.
Then I measured the conductivity of each solution using graphite electrodes as detailed in the section above. I used a voltage of 4.00 V AC and recorded the currents in milliampere (mA) as shown in the table. I made sure I used the AC scale on the voltmeter and ammeter.
RESULTS. The table below also shows the [H+] as calculated from the Ka value of 1.80 x 10-5. The formula is:
To calculate the total ionic concentration you just double the [H+] value. The conductance is C = I/V and has the units siemens, S. I converted this to microsiemens µS. I also know the surface area of the electrodes (A = 4 cm2) and the distance apart (L = 0.90 cm). This enables me to calculate conductivity, κ, in µS per cm. The formula is: κ = I/V x L/A. For very, very dilute solutions you may need to use the microampere (µA) scale on the ammeter.
Conductivity, κ (µS cm-1)
ANALYSIS. I've plotted two graphs as shown below:
HINTS. 1. Use an AC power supply. This gives a more consistent reading for current. But make sure the ammeter and voltmeter are both set to read AC. 2. Keep voltage constant. Voltage will change depending on the solution and you will need to be able to adjust the voltage accordingly. You can't just set your power supply to 6 V and think it will not change. Just record both the volatge and current a the same time. See my notes in the previous section. 3. Choose the right voltage. Try out your most concentrated solution to see what the maximum voltage is that your power supply can deliver. It may be able to give 10 V for a dilute solution but only 4 V for a concentrated one, so choose 4 V for all.
Here's a great experiment that is simple to do and gives great results in a short time. We can get an experimental value for the acid dissociation constant Ka and compare it to the accepted or true value.
The pH of a strong acid like hydrochloric acid or nitric acid is fairly straightforward; it is just the −log10[H+], and because strong acids fully dissociate, the [H+] at equilibrium is the same as the initial concentration of the acid itself. So, when we talk about 0.1 M HCl we are really saying [HCl] = 0.1 M initially, and that [H+] = 0.1 M at equilibrium. That is:
However, for weak acids such as acetic acid, CH3COOH, this is not true. In that case we factor in the acid dissociation constant, Ka, and use the approximation:
• distinguish between strong and weak acids and bases in terms of the extent of dissociation • distinguish between the terms strong and concentrated for acids and bases • understand that the pH scale is a logarithmic scale • Suggested practical: Measure pH of a substance.
Chemistry 2025 v1.1, General Senior Syllabus, Queensland Curriculum & Assessment Authority July 2024, page 40
My suggestion is to make up a series of acetic acid solutions of varying concentrations and measure the pH of each. We can then see if the equation works and what experimental value we get for Ka.
My results. I recorded the pH of each solution, and from that calculated the [H+] concentration by using the relationship [H+] = 10-pH. I know from the formula that [H+] will be proportional to the square root of the initial concentration:
So, I've also calculated the square root of the initial concentration of CH3COOH ready for plotting:
I've plotted both [H+] vs [CH3COOH], and [H+] vs square root of [CH3COOH]:
From the linear graph (on the right) we can make use of the gradient to determine the acid equilibrium constant, Ka, for acetic acid. The calculations are shown below. Firstly, I've rearranged the middle equation above to show how to use the gradient to calculate Ka:
The experimental (observed, or measured) value of Ka of 1.78 x 10-5 is quite close to the accepted (true) value of 1.80 x 10-5, and so we can work out the absolute error (Ea = 1.78 x 10-5 − 1.80 x 10-5 = −0.02 x 10-5). To work out percentage error we use the following formula:
This value is quite low and it means the experiment was quite valid. I tell my students that a percentage error of less than 5% is acceptable for senior chemistry classroom experiments.
This type of experiment could be used for other weak acids such as citric or oxalic acids. Oxalic is great because it is a primary standard and you can make up a standard 1M solution of oxalic acid by weighing out the exact amount of dry powder and making it up to the required volume, and then diluting as needed. Alternatively, you could just standardise it against standard NaOH solution. Be careful though as oxalic is diprotic, and citric is triprotic and that complicates the calculations. You could always use the weak acid potassium hydrogen phthalate (KHP) as it is a primary standard and monoprotic.
Here's a variation on the experiment above using different graphical analysis. It is an interesting approach using a new set of data with uncertainty for the pH shown using error bars.
The pH is related to concentration of a weak acid such as acetic acid in the following way:
This means if we plot pH (y-axis) against log10[HAinitial] on the x-axis we should get a linear graph with a gradient of −0.5 and a y-intercept of −2.4
MY EXPERIMENTAL METHOD
I made up six solutions of acetic acid of different concentrations. I just started with "Double strength vinegar" from Coles. This is 8% acetic acid according to the label, and I determined its concentration by titration against standard 0.1 M NaOH using phenolphthalein indicator. I has three concordant titres and that gave a value for the vinegar of 1.30 M acetic acid. From this I did 5 serial dilutions by taking 50 mL of the 1.30 M acetic and diluting it to 100 mL with deionized water to produce 0.65 M acetic. I kept 50 mL of this for the experiment and diluted the remaining 50 mL to 100 mL to produced 0.325 M acetic. Again, I kept 50 mL of this and diluted the remaining 50 mL to 100 mL with water to produce 0.16 M acetic. And so on.
The best way to measure the pH is to start with the most dilute and work towards the most concentrated. That way you are not affecting the concentration of the samples as you go. Once you've do one set (Trial 1) rinse the probe in water and then start Trial 2. And so on.
MY EXPERIMENTAL RESULTS
When you plot pH vs concentration you will get a logarithmic relationship. Here's mine based on the results above. Note that the equation for the trendline is indeed logarithmic (ln).
When I plot pH vs log of concentration the graph is linearised and we get a linear graph with a negative gradient.
I've added error bars based on the uncertainty in the pH results (see δx̄ in the table above).The equation for the trendline (dotted blue line) is shown on the graph as y = 0.4707x + 1.6789 with an R2 = 0.9984
UNCERTAINTY IN THE GRADIENT The equation for the maximum trendline (green) is y = 0.55x + 1.64, and the equations for the minimum linear trendline (orange) is: y = 0.37x + 1.74. From these two equations we can work out the uncertainty in the gradient by calculating their difference and dividing by 2. We get (0.55 − 0.37)/2 = 0.09 pH units. This is a percentage uncertainty δ%̄ = 20% (to 1 d.p.). The range for the experimental value for the gradient is therefore 0.4707 ± 0.09 or from a low of 0.38 to a high of 0.56 units.
UNCERTAINTY IN THE Y-INTERCEPT This can be calculated from the equations in the same way and we get (1.74 − 1.64)/2 = 0.05
EQUATION FOR THE TRENDLINE INCLUDING UNCERTAINTY The full equation can be expressed using absolute values for the uncertainty (δx) in the gradient, or with percentage uncertainty (δ%): y = (0.47 ± 0.09)x +(1.68 ± 0.05) y = (0.47x ± 20%) + (1.68 ± 0.05)
They can also be expressed using the actual (S. I.) symbols for the quantities (note that you never convert the uncertainty in the intercept to percentage as you can get infinite values). I've used "C" to represent the initial concentration of the acetic acid. pH = (0.47 ± 0.09) logC + (1.68 ± 0.05) pH = (0.47 ± 0.09) logC + (1.68 ± 0.05)
ERROR IN THE GRADIENT AND Y-INTERCEPT The accepted (true) value for the gradient was stated before as 0.50 and I've got a value of 0.47. That's an error of 0.03 or 6%. This means the prac is a fairly accurate way of measuring the gradient.
DOES THE EXPERIMENTAL RANGE OVERLAP WITH THE ACCEPTED VALUE? The accepted value of 0.50 is within the experimental range of 0.38 to 0.56 units, so we can say that within the uncertainty of our experimental results our value is quite reasonable. However, it is the accuracy of 6% that is more important, and that is also quite good. What a great prac!
6. HOW DOES THE pH OF WATER VARY WITH TEMPERATURE?
The pH of water will decrease as the temperature increases. This makes a great investigation and is very easy and quick to do. You can do it in 4 minutes, so that's 12 minutes of data collection time for the whole prac including replicates. And it works so well!
The Queensland syllabus lists the following points about the equilibrium in water:
Understanding• Determine the effect of temperature change on chemical systems at equilibrium by considering the enthalpy change for the forward and reverse reactions. • Apply Le Chatelier's principle to determine the effect changes of temperature (...) has on the position of equilibrium and on the value of the equilibrium constant. • Identify that water is a weak electrolyte and the self-ionisation of water is represented by Kw = [H+][OH-].
Investigate • factors that affect equilibrium (Le Chatelier's principle) QCAA Chemistry General Syllabus - 2025 v1.0 page 35.
Water molecules undergo an endothermic dissociation: H2O (l) + energy ⇄ H+(aq) + OH–(aq), so when temperature is increased the equilibrium will shift to the right and the concentration of both H+ and OH- will be increased. An increase in [H+] means the pH gets lower. This makes a great student experiment. However, it is not a negative linear relationship between pH and temperature but, according to theory, it should be an inverse one. That is, pH is proportional to 1/T.
THEORY. The Van 't Hoff equation can be applied to this equilibrium. The variable ln Keq is proportional to pH and now we can see that pH is proportional to the reciprocal of temperature:
THEORETICAL CALCULATIONS. We know that the equilibrium constant for water Kw at 25°C is 1.00 x 10-14. This is stated in the QCAA Formula and Data book as:
The units are the same as (mol/L)2. We also know from the Formula book that: Kw = [H+][OH-], and as [H+] = [OH-], we can say that at 25°C (296 K) the value of [H+] = square root of Kw which equals 1.00 x 10-7 M. This gives us a pH at 298 K of 7.00 units. To determine the pH of water at other temperatures we need to use the Van 't Hoff equation rearranged into this form:
Use that method in Excel to calculate the theoretical pH of water at any temperature. Note that you need to keep T1 as 298 K, and K1 at 10-14. Here are my calculations:
And here are the graphs from the table:
Graph of pH versus 1/T. This is a great fit (R = 1.00) and is in agreement with calculations from the the Van 't Hoff formula. This can be regarded as the accepted value for the gradient (1495.8) and y-intercept (1.9831) when calculating error for your own experiment.
MY EXPERIMENT
THE WATER. Tap water is not suitable for this experiment as I quickly found. The pH of tap water is somewhere between 6.5 to 8.5 but that is not the problem. Tap water had many additives that prevent the equilibrium responding as you'd expect if it was just water. It has natural chemicals present such as calcium and magnesium carbonates and bicarbonates, as well as added chemicals such as calcium hydroxide and hydrochloric acid. These will act as buffers and stop the pH of water changing as you'd expect. The change in pH is much lower and the experiment is not as successful.
DEIONISED WATER IS BEST. My recommendation is to used deionised water (or distilled water). However, any water will absorb carbon dioxide from the atmosphere and the pH will be lower than 7.0 so you need to control exposure to CO2. I found that boiling water for a minute and then letting it cool to room temperature in a sealed container is best. A glass bottle with a lid is fine. When testing, take a sample (100 mL) in a beaker and reseal the bottle. SETUP. Here's my setup using a pH meter that has automatic temperature compensation (ATC). I just put the pH meter in a beaker of water and placed it on a hotplate. I turned it on and recorded temperatuer and pH data. Actually, I just videoed it and extracted the data later. I found that using a magnetic stirrer gave more even results and I would recommend that. Be aware that if your pH meter doesn't have a built-in thermometer it may not compensate automatically for the effect of temperature and you will need to do this manually. It may give you exaggerated readings. The specifications that come with the meter should tell you.
Here's my graph:
COMMENT TO STUDENTS. You may like to plot pH vs T to start with and then try linearising it by plotting pH vs 1/T. In Senior Chemistry it is not expected that you linearise graphs as I did, but you can comment on the shape and compare the gradient and intercept to my theoretical one earlier. That will help you establish the accuracy of the experiment. However, if you do linearise it you are then able to calculate the experimental value of the heat of dissociation (ΔH) and compare it to the accepted value for ΔHo of 57.270 kJ/mol.
CALCULATING ΔH. The gradient of the line of best fit (the dotted blue line) is 1640.7 and the y-intercept is 1.5267 units. A value for R2 is high which indicates the equation for the line of best fit is a good fit for the data. The experimental value for ΔH is the gradient of the pH vs 1/T graph multiplied by a constant of 38.29, thus:
ERROR ANALYSIS. I can now calculate the absolute error (Ea) and percentage error (E%) for my experimental results:
UNCERTAINTY AND PRECISION OF THE GRADIENT. In Senior Chemistry it is not usual to calculate the unceratinty of results using the graph. We do this in Senior Physics but I'll show you how it is done. You'll note that on the graph I have added error bars to show the uncertainty of each data point. The I've drawn a maximum gradient trendline (green) which fits between ALL the error bars, and I've drawn a minimum gradient trendline (red) which also fits between ALL the error bars. Then I've calculated the uncertainty in the gradient by subtracting the minimum gradient from the maximum and divided by 2:
Maximum trendline gradient = (7.22-6.18)/(0.00342-0.0029) = 2000 Minimum trendline gradient = (7.1-6.4)/(0.00342-0.0029) = 1346 Absolute uncertainty in the gradient = (2000 - 1346)/2 = 327 Percventage uncertainty in the gradient = 327/1640.7 x 100 = 20%
From this we can determine the range of values for our experimental ΔH. It is just 62882 J/mol ±20% or 62882 ± 12576. This is a range of 50306 to 75387 J/mol.
CAN WE SAY OUR RESULTS ARE PRECISE? An uncertainty of 20% is quite high but what we can now do it to see if the range of results includes the accepted value. If it does we can say that the experiment was valid (accurate) as the accepted value is in the range of experimental results given the uncertainty of the experiment. The accepted value of 57270 J/mol is within the range of 50306 to 75387 J/mol. What a great result.
HOW DO WE STATE THE RELATIONSHIP INCLUDING THE UNCERTAINTY? Like this:
pH = (1640.7 ± 327) T + 1.5267 pH = 1640.7 T ± 20% + 1.5267
There are far too many significant figure in this equation so we should round it off to just 3:
pH = (1640 ± 327) T + 1.53 pH = 1640 T ± 20% + 1.52
THEORY BEHIND VAN 'T HOFF EQUATION AND PH (THIS IS NOT NEEDED FOR YOUR REPORT)
The Van 't Hoff equation can be developed further to calculate pH of a solution. We start with the Van 't Hoff equation (Wikipedia):
WHAT ABOUT CHANGE IN ENTROPY? Entropy is not a part of the Queensland physics syllabus, nevertheless, we can still see how the calculation goes. Enthalpy is a measure of the degree of disorder or randomness of a system. High entropy means high disorder. When a water molecule dissociates into its ions it goes from one particle to two particles so it is said to increase in entropy. According to Engineering Toolbox, the standard entropy of water at 20oC is 5.341 J/mol and at 60oC is 14.98 J/mol. This is consistent with the notion that entropy increases. The change in standard entropy ΔSo = 14.98 − 5.341 = +9.64 J/mol. It is a positive numbre indicating that entropy has increased.
ERROR IN CHANGE IN ENTROPY The experimental change in entropy is given by the y-intercept of the graph above. ΔSo = y-intercept x 38.29, that is 1.5267 x 38.29 = 58.46 J/mol. The absolute error is 58.46 − 9.64 = 48.82 J/mol. This is way out but at least it is positive as expected. Maybe I have the wrong theoretical value. That's all I can think of. Good luck!
7. HOW DOES THE pH OF ACETIC ACID VARY WITH TEMPERATURE?
The pH of acetic acid will decrease as the temperature increases. This makes a great investigation and is very easy and quick to do. You can do it in 4 minutes, so that's 12 minutes of data collection time for the whole prac including replicates. And it works so well!
The Queensland syllabus lists the following activities under Science Inquiry that are relevant to our experiments on acetic acid:
Investigate: • factors that affect equilibrium (Le Chatelier's principle) • properties of acids and bases QCAA Chemistry General Syllabus - 2025 v1.0 page 35.
You may be aware that when substances dissolve in water the solution may get hot or cold. This is called Heat of Solution, ΔHsoln. Acetic acid is an acid because it's -O-H bond can break, donating a hydrogen ion to whatever's around to accept it - mostly forming H+(aq) (written as H3O+). With more energy in the solution at higher temperatures the -O-H bond is broken more readily. So the higher the temperature the more dissociated the acid and the lower the pH. The same is true of most, if not all, carboxylic acids.
The first reaction to occur when acetic acid dissolves is the bond breaking of the O-H bond in the -COOH functional group. Bond breaking is endothermic as it requires energy:
CH3COOH(l) + energy → H+ + CH3COO–
The second reaction is between the hydrogen ion and a water molecule. This is bond making so is exothermic:
H+ + H2O → H3O+(aq) + energy
CH3COOH(l) + H2O + energy → H3O+(aq) + CH3COO–(aq)
In terms of Le Chatelier's principle, when a solution of acetic acid is heated, the added energy shifts the equilibrium to the right and the concentration of H+ ions in solution increases, and thus the pH decreases. This forms the basis of a good student experiment.
THEORY - THE VAN 'T HOFF EQUATION To calculate the theoretical pH for an acetic acid solution at various temperatures we need to use the Van 't Hoff equation:
It enables us to find the equilibrium constant (Keq, or Ka) based on values for heat of solution ΔHo and change in entropy ΔSo at a particular kelvin temperature, T. You can find this equation in Wikipedia for example. It relates the natural log of the equilibrium constant (ln Keq) to the temperature of the solution. For acetic acid, the heat of solution, ΔHo, is 1.2 kJ/mol (1200 J/mol), and the term ΔSo is called the change in entropy which has a value of -86.9 J/mol. The R term is the gas constant equal to 8.314 J/mol/K. When we substitute the values in at a temperature of 298 K (25oC) we get ln Keq equal to -10.937. Raise the natural base e to this value and we get 1.779 x 10-5. This is the Keq value which we refer to as Ka when it is an acid like acetic acid. From that you can determine [H+] and pH (= 2.38) in the usual way.
We can repeat this for other temperatures and that will show us the effect of temperature on pH. For example, here's the same calculation as above but for a temperature of 323 K (50oC):
The theory comes from the Van 't Hoff equation which suggests that the natural log of the acid equilibrium constant, ln Ka, is proportional to 1/T so we should plot that and it should be linear:
MY EXPERIMENT To test this out I used a pH meter in a solution of 0.50 M acetic acid. I just used commercial cleaning vinegar from Woolworths ($3/2L) which is a 5% solution of acetic acid in water. It has a concentration of 0.86 M, so I diluted it to 0.50 M and standardized it against 0.100 M NaOH using phenolphthalein indicator. You don't need to dilute it though, but you should know its concentration.
My experimental results for 0.5 M acetic acid. This was done without stirring.
My experimental results for 1.0 M acetic acid. This worked so well that I repeated the experiment using a hotplate with a magnetic stirrer. I also used 1.0 M acetic acid (standardised against standard 0.100 M NaOH solution). The experiment was done three times to minimize random error and improve the precision of the result. Here's my graph:
COMMENT TO STUDENTS. You may like to plot pH vs T to start with and then try linearising it by plotting pH vs 1/T. In Senior Chemistry it is not expected that you linearise graphs as I did, but you can comment on the shape and compare the gradient and intercept to my theoretical one earlier. That will help you establish the accuracy of the experiment. However, if you do linearise it you are then able to calculate the experimental value of the heat of dissociation (ΔH) and compare it to the accepted value for ΔHo of 1200 J/mol.
CALCULATING ΔH. The gradient of the line of best fit (the dotted blue line) is 2356 and the y-intercept is 5.7 units. A value for R2 is high which indicates the equation for the line of best fit is a good fit for the data. The experimental value for ΔH is the gradient of the pH vs 1/T graph multiplied by a constant of 38.29, thus:
ΔH = gradient x 38.29 = 2356 x 38.29 = 90211 J/mol
ERROR ANALYSIS. I could now calculate the absolute error (Ea) and percentage error (E%) for my experimental results but the experimental value of 90211 J/mol is abut 7 times too great. The error would be enormous so I suspect the value of 1200 J/mol I used as the accepted value is not the right value.
Maximum trendline gradient = (2.22-1.25)/(0.00336-0.00296) = 2425 Minimum trendline gradient = (2.18-1.27)/(0.00336-0.00296) = 2275 Absolute uncertainty in the gradient = (2425 - 2275)/2 = 75 Percventage uncertainty in the gradient = 75/2356 x 100 = 3.2%
From this we can determine the range of values for our experimental ΔH. It is just 90211 J/mol ±3.2% or 90211 ± 2887. This is a range of 87324 to 93098 J/mol.
CAN WE SAY OUR RESULTS ARE PRECISE? An uncertainty of 3% is excellent but what we can now do it to see if the range of results includes the accepted value. If it does we can say that the experiment was valid (accurate) as the accepted value is in the range of experimental results given the uncertainty of the experiment. The accepted value of 1200 J/mol is not within the range of 87324 to 93098 J/mol so we must conclude that the experiment is not accurate. It is not a valid way to measure the heat of solution based on the accepted value of 1200 J/mol.
pH = (2356 ± 75) T + 5.7 pH = 2356 T ± 3% + 5.7
JASMINE AND HANNA'S RESULTS Here are some results from students Jasmine and Hannah from Calvary Christian College who used 1.0 M acetic acid and a Vernier pH sensor that sampled the pH every 2 seconds using a datalogger. They used a digital thermometer for temperature results.
Here's their setup:
And here are graphs of their results:
ERROR ANALYSIS. Here's a way of working out uncertainty in the gradient using maximum and minimum trendlines.
We can calculate the value of ΔH = 1418 x 38.29 = 54320 J/mol. If we assume the heat of dissociation is 1200 J/mol then we have a 53% error. The value of ΔS is more accurate. You get 2.2857 x 38.29 = 87.6 J/mol which is an absolute error of 0.016 J/mol, or 1.9%. That is amazingly good.
You can't use a titration to measure the effect of temperature on the dissociation of a weak acid. Let me explain. Students often say that a change in temperature will shift equilibrium to increase the amount of H+ ions, so the pH will decrease with increasing temperature. Students then say they will measure this changed acidity by doing a titration. THIS WILL NOT WORK. No matter what temperature the solutions are at they will all have identical titres. It is true that there won't be as many H+ ions in solution before the titration begins, however, as the titration proceeds these H+ ions are used up by the OH- ions from NaOH to form water. By Le Chatelier's Principle, the equilibrium shifts to the right, and more H+ ions are then released by the molecules. But these too are used up by the NaOH in the titration. By the end point all molecules have dissociated and all H+ have reacted. So, in a titration every molecule will have dissociated by the end point. That is, 0.1 mol of acetic acid will form 0.1 mol of H+ ions no matter what temperature it is at; and 20 mL aliquots of 0.100 M acetic acid will have the same titre no matter what temperature they are at.
If you don't believe me, here are my results for 20.0 mL aliquots of 0.100 M acetic acid titrated against 0.155 M NaOH, at various temperatures. The temperatures are averages of the starting and final temperatures. The temperature drops as the room temperature NaOH is added. For example, when I used acetic acid at 80°C and NaOH at 25°C it finished up at 60°C, so the avearge temperature in the flask was 70°C.
See, no trend in the titres. I'd be really concerned if there was one.
(b) Conductivity change. According to Le Chatelier's Principle, as you increase the temperature the concentration of ions is reduced because equilibrium shifts to the left. When I tried heating a sample of 0.100 M acetic acid I got the following values for current and conductivity. I have shown a graph of the results.
It does seem that conductivity of acetic acid doesn't decrease with an increase in temperature. In fact it goes in the reverse direction - conductivity increases with temperature. This is easily explained by the fact that conductivity of most ionic solutions increases with temperature. This isn't because there are more ions present - in fact there are fewer ions as we know from equilibrium shift to the left (as described earlier). What happens is that hydrogen-bonding between the solvated ions (H+-H2O) is weakened by the added energy and breaks them apart. This means the ions are not so strongly attracted to each other so are more mobile. This means conductivity increases.
All we know is that as temperature increases, there are fewer ions because of the equilibrium shift, but they are more mobile. The nett result is an increase in conductivity. We just can't make any statements about the nature or size of the equilibrium shift as it is hidden by the decrease in H-bonding.
This can still make a good student experiment but it doesn't say much about the equilibrium position of a weak acid when it is heated. It just says the tested ionic substances, whether weak or strong acids, or non-acids, increase in conductivity with temperature.
The titration of hydrochloric acid with sodium hydroxide is exothermic, so as the titration proceeds, the temperature rises until the end point. This is called a thermometric titration. They are not new and go back to 1913. After the end point, as more sodium hydroxide is added the temperature starts to go down because there is no further reaction and you're just adding cool sodium hydroxide to a flask that is warmer. The graph is curved with the end point being at the point of highest temperature.
SYLLABUS. The Queensland chemistry syllabus lists the following Unit 2 subject matter:
However, most students will be doing their student experiment in Unit 3 (Equilibrium, acids and redox reactions), so the following subject matter is more relevant:
• use appropriate mathematical representations and analyse experimental data and titration curves to solve problems and make predictions, including using the mole concept to calculate moles, mass, volume and concentration from volumetric analysis data. • Mandatory practical: Acid-base titration to calculate the concentration of a solution with reference to a standard solution. Chemistry 2019 v1.4, op. cit. p 42.
A good experiment that fits in well with these points is measuring the temperature change during a titration.
AIM. To test the accuracy of a thermometric titration in determining the end point of an acid-base titration
To test this I measured the temperature of HCL/NaOH reaction mixture in an insulated cup as a function of volume of sodium hydroxide added from the burette. I used 20.0 mL of 1.00 M HCl and titrated it against 1.00 M NaOH from the burette. I also used a digital thermometer but a glass one would be okay. The reaction is exothermic and the Heat (Enthalpy) of Neutralisation, ΔH, is 57.460 kJ/mol. It can be written like this:
At the start: T = 21.8°C V (HCl) added = 0 mL
Here's my graph. The temperature reading was constant at 26.4°C from 19.5 mL to 20.5 mL, probably due to the scale reading uncertainty of the thermometer (±0.1°C). The indicator (phenolphthalein) changed colour at 20.0 ± 0.1mL. Note: the scale reading uncertainty for a burette is ± 0.05 mL (half-scale division) for the initial burette reading (IBR) and another ± 0.05 mL for the final burette reading (FBR), giving a total uncertainty for the titre of ± 0.1 mL. The tip of the graph is called the 'inflection point' or the 'breakpoint' and indicates the end point.
The exact location of the inflection point is not easy to determine when it is not a sharp peak in temperature. In the graph above it looks like it is somewhere between 19.5 mL and 20.5 mL but somtime you get a much wider range. I've had it range over 2 mL which seems very broqd compared to using an indicator like phenolphthalein which changes colour over 1 drop of base (0.05 mL). The usual way to narrow it down is to draw straight lines of best fit on each side and noting where they intersect.
THEORETICAL RESULTS: We can determine the theoretical value of temperature at any point in the titration. It is a matter of applying calorimetry principles.
EXAMPLE 1: Half equivalence point (the buffer region). Question 1: Calculate the temperature of a mixture of 20.0 mL of 1.00 M HCl at 21.8°C and 10.0 mL of 1.00 M NaOH at 21.8°C. Solution 1: We need to work out the number of moles of each reactant. n(HCl) = C(HCl) × V(HCl) = 1.00 × 20.0/1000 = 0.0200 mol n(NaOH) = C(NaOH) × V(NaOH) = 1.00 × 10.0/1000 = 0.0100 mol The stoichiometric ratio is 1:1, so NaOH is the limiting reagent. Thus, the number of moles of reactants used is 0.0100 mol.
EXAMPLE 2: Equivalence point. Question 2: Calculate the temperature of a mixture of 20.0 mL of 1.00 M HCl at 21.8°C and 20.0 mL of 1.00 M NaOH at 21.8°C. Solution 2: We need to work out the number of moles of each reactant at the end (equivalence) point. n(HCl) = C(HCl) × V(HCl) = 1.00 × 20.0/1000 = 0.0200 mol n(NaOH) = C(NaOH) × V(NaOH) = 1.00 × 20.0/1000 = 0.0200 mol The stoichiometric ratio is 1:1, so n(NaOH) will equal n(HCl). Thus, the number of moles of each reactant used is 0.0200 mol.
EXAMPLE 3: Twice the equivalence point Question 3: Calculate the temperature of a mixture of 40.0 mL of the reaction mixture at 28.6°C and 20.0 mL of 1.00 M NaOH at 21.8°C. Solution 3: There is no further reaction so no more heat of neutralisation is released, so we need to use a method of mixtures. By the Law of conservation of energy:
COMPARISON OF RESULTS: We can plot the theoretical and experimental graph together:
ACCURACY OF RESULTS. It is difficult to draw a conclusion about the accuracy of these results because there is no accepted value to compare our results to. This is not a thermochemistry experiment in the sense that we are interested in the value for the Heat of Neutralistation. We do see though that the maximum temperature (at the end point) is the same for the theoretical value and experimental value, so that confirms the accuracy of the enthalpy calculations. But we are more interested in whether the maximum temperature is reached at the end point as theory would suggest. We can see from the experimental graph that the maximum temperature of 28.6°C occurs over the range of 19.5 mL to 20.5 mL which confirms the results, although with an certainty of 0.5 mL (2.5%). This is quite an acceptable accuracy and we can say confidently that our results confirm the theory used. I suspect the difference in the second part of the graph is due to loss of heat through the surface. Why this didn't happen on the heating side of the graph I do not know.
WINE AND SOFT DRINK TITRATION EXPERIMENTS
Fermentation and the alcoholic content of wine - analysis by titration
The quality of Queensland wines is now recognised as amongst the best in Australia. Overseas exports are increasing, particularly to international markets seeking premium quality boutique wines. The Queensland wine industry has grown significantly over the years to cover a total of 1400 hectares. The majority of this growth has occurred during the past 20 years with significant plantings throughout the southeast corner of the State. However, winemaking is still somewhat of an art but is strongly informed by science. Thus an interesting student experiment can be undertaken in this context.
THE MOST IMPORTANT INDEPENDENT VARIABLES IN FERMENTATION
There are two key independent variables worth considering:
(a) Sugar concentration. After crushing the grapes the next step in the making of wine is the fermentation of the grape juice and pulp with various yeasts and bacteria. Most books say that the amount of ethanol produced is dependent on the sugar concentration of the starting juice but then give four different equations depending on assumptions made (such as the ratio and purity of glucose and fructose; or whether the fermentation gets 'stuck' at the primary stage). The most common relationship is a linear one (y = 0.6x -1) - so an EEI could investigate that.
A good student experiment would be to use fresh grape juice or simulate grape juice with 150-250 g/L glucose (or an equal mix of glucose and fructose), adding a controlled amount of yeast and wine acids and fermenting to stillness at constant temperature.
(b) Temperature. Fermentation is an exothermic reaction so heat is generated during the process. To control the heat, the winemaker must choose a suitable vessel size or else use a cooling device. In the case of a student experiment , you might control the temperature by use of a water bath (or a refrigerator). Typically, white wine is fermented between 18-20°C though a wine maker may choose to use a higher temperature to bring out some of the complexity of the wine. Red wine is typically fermented at higher temperatures up to 29°C. Fermentation at higher temperatures may have adverse effect on the wine in stunning the yeast to inactivity and even "boiling off" some of the flavors of the wines. How temperature and the final concentration of alcohol are related would make an ideal student experiment.
OTHER INDEPENDENT VARIABLES WORTH CONSIDERING
(c) Acidity. This is not such an important one and the effects may be small - but nevertheless important. You could repeat it with acidity as the independent variable and controlling the amount of sugar, yeast, temperature and so on.
(d) Type of yeast. As the alcohol concentration rises the yeast cell membranes become susceptible to rupture by the ethanol. Some yeasts are more susceptible than others. Baker's yeast is very susceptible and will die at just a few % alcohol; brewer's yeasts (for beer) are okay up to 5% but some can survive in up to 9% alcohol; and wine yeast usually go from about 13% (Sav Blanc), Riesling (16%) and a sherry yeast can tolerate about 17%. Or you could look at the susceptibility of yeasts to [SO2] - winemakers use SO2 in the form of sodium metabisulfite to kill off wild yeasts as these are less tolerant than wine yeast to the SO2. The interesting thing is that you could breed a yeast to survive higher alcohol concentrations (like they do at wineries for their malo-lactic fermentation of sparkling wines) by slowly increasing the amount of alcohol in the brew from say a few % to 10% over a period of a week so that the yeast adapts. This sounds more like a Biology EEI so I'd better stop here.
(e) Sulfur dioxide. Sulfur dioxide is widely used in winemaking because of its antioxidant and antibacterial properties. You could hypothesise and test how SO2 affects the performance of yeast. Two common methods for determining SO2 in wine are included in the "sulfite" section later.
SCHOOL WINEMAKING IN GENERAL
Any fruit (or juice) works just fine although some require more sugar to be added. James Palframan HOD Science at MacGregor High School Brisbane adds a salutary note: "My year 12 chemists last year made a range of wines including lychee, lemon, ginger, dragon fruit, apple and mango. The dragon fruit wine ended up being very expensive at approximately $10 to $12 a bottle as the dragon fruit was quite expensive to start with and then the students discovered when that chopped it up that it was mostly water and not a lot of fruit pulp". Two successful used Wine EEI task sheets are from Otto Craig Wine EEI #1" and Melissa Dixon Wine EEI #2 (but remember, these are not exemplars, they are just from submissions that have been to QSA District or State Panels for the purpose of review). Also, my winemaking unit for chemistry teachers is available online.
It is worth stressing here that you should not taste the wine being produced; this is not because of the alcohol but rather because of the unsanitary conditions under which your wine is being made (in a lab, not the Home Economics kitchen). If you intend tasting your wine then your risk assessment should state and evaluate that. If I was your teacher I'd say "no" as it would not be worth the headache.
THE DEPENDENT VARIABLE MEASURING THE ALCOHOL CONCENTRATION IN WINE
The most common method is by redox titration. In this analysis, you add an excess of standardized acidified potassium dichromate solution to the wine which converts the ethanol to ethanoic (acetic) acid. The amount of unreacted dichromate is then determined by adding an excess of potassium iodide solution which is also oxidised by the potassium dichromate to form iodine. The iodine is then back-titrated with a standard solution of sodium thiosulfate and a starch indicator. The titration results are used to calculate the ethanol content of the original solution. It is complex but works well and is very impressive.
You have a problem if you are dealing with red wine as the red pigments mask the colour changes. In that case you have to extract the ethanol from the wine (in effect, by various forms of distillation) and carry on, as above, from there. Canterbury University NZ has quite a simple method for red wine: see Canterbury - alcohol titration. Chemistry teacher Emma Hodginkson from Mountain Creek State High has performed the Canterbury ethanol titration with her Year 12s for a few years and has found it very successful. When analysing commercial wine, her students get very close to the % alcohol on the label. As it is a redox titration she says it works better in Year 12 when the students have completed some redox theory whereas Year 11s find the calculations a bit heavy going. To overcome the practical difficulty in locating a small container to suspend above the dichromate - they use a plastic water bottle lid suspended with cotton thread. Cool!
At All Hallows' School, Brisbane, chemistry teacher Matthew Stuart uses a more compact method: a boiling tube is used to hold the 20 mL wine sample, and 8 mL dichromate solution is placed in a small 'fusion tube' (a small diameter test tube) carefully slipped into the wine sample. Some parafilm is used to seal the boiling tube, or a suitably sized stopper if possible. With care, the small inner test tube will float on top of the wine sample, and these are left in a drying oven (50°C) overnight to drive (evaporate) the ethanol to the dichromate solution. A pair of forceps is used to remove the dichromate tube without spilling into the wine sample, the outside rinsed with water, and then titrated. One caution in all of this: merely measuring the concentration of the various components of a selection of wines (ethanol, pH, acidity and so on) may not make a good student experiment. Manipulation of variables gives students a better chance of demonstrating all aspects of the assessment criteria.
1. Alcohol content by distillation 2.Alcohol content by Ebulliometry 3. Alcohol content by boiling 4. Alcohol concentration by dichromate oxidation and titration 5. Alcohol concentration by Vinometer 6. Alcohol concentration (Western Australia - Chemistry in Context)
Yeast type and alcohol concentration in wine
You may have seen different types of yeast for different purposes; for example, there are beer yeasts (top-cropping ale strains and bottom-cropping lager strains); baker's yeast for bread-making; red and white wine yeasts; and even genetically engineered yeasts used for industrial alcohol production. Many are just different strains of the same yeast but grow differently. A good research question might be: how do different yeasts affect the production of alcohol from grape juice?
There is a problem however. The majority of senior chemistry investigations use 'continuous' variables; that is, any value is possible within the limits the variable ranges. For example, temperature, pH, sugar concentration, amount (mass) of yeast, surface area and so on. But when you are comparing two different categories of a variable, such as type of yeast (wine yeast vs. baker's yeast) then you have a categorical variable. The choice of the categorical variables is not as common as using a continuous variable in Senior Chemistry experiments, partly because categorical variables have their peculiar difficulties and this makes the design of this EEI far more complex than it looks at first glance. It is not just a matter of comparing equal amounts of the two yeast products on the amount of alcohol produced.
A decision has to be made about the amount of each product to use to get some sort of equivalent mass of yeast for comparison (and how this is arrived at; is there any indication of the % composition of the two products). Do the yeasts each have an optimum pH and if so what pH will be chosen for the grape juice (and why)? I know that the Lalvin BGY yeast from Burgundy, France is hopeless at pHs lower than 3.2 but other work at higher pHs. Is surface area a concern (maybe if one is a bottom fermenter, and another a top fermenting yeast). What temperature will be used (and why) if the yeasts have their own optimum temperature for growth; for example the BGY Lalvin yeast from Burgundy, France works best at 24°-28°C. Will a low sugar or high sugar juice be used - important as it may be the alcohol itself that inhibits the yeast. For example, the Lalvin CLOS yeast from Spain is high-alcohol-tolerant up to 15% alcohol but others give up before that. And what about the dependent variable (alcohol concentration): will the rate of alcohol production be measured, or just the amount of alcohol present when the yeasts die or the sugar runs out; or will the alcohol be measured after a set time, eg 7 days? Is time important? Some yeasts are slow (eg the CY3079 Slow White yeast from France takes its time but gets there in the end; it would be a brave decision to cut it off after 7 days). Lastly, some yeasts convert malic acid to alcohol (as well as converting the sugar). Imagine using a yeast such as the Lalvin C from France which partially degrades malic acid. Of course you'd get more alcohol out of this one.
Which ferments best: glucose, fructose or sucrose?
Another terrific idea for a wine student experiment. The two common fruit sugars used in winemaking are glucose and fructose. Grape juice is made up of these in roughly equal quantities. Another sugar used in the fermentation industry is sucrose (cane sugar). Sucrose is frequently used as a cheap carbohydrate by breweries, wineries and other fermentation-based industries employing yeast. It is a di-saccharide composed of D-glucose and D-fructose linked by an alpha-1,4 glycoside bond. In the initial stages of fermentation, sucrose is rapidly hydrolyzed into glucose and fructose by the action of the enzyme invertase on this bond. Then the sugars are transported across the cell membrane where they ferment and form alcohol. So investigating the fermentation of sucrose is really also about studying the fermentation of a mixture of glucose and fructose. This suggests a terrific student experiment: to look at the reaction rates of glucose, fructose and sucrose. I am indebted to Savannah from Coolum State High School for suggesting this to me.
Glucose fermenter. May 2014.
At the start of fermentation, grape juice contains approximately equal amounts of glucose and fructose (called 'hexose' sugars). While both are fermented by wine yeasts to ethanol and carbon dioxide, Saccharomyces cerevisiae consumes glucose faster than fructose because this yeast is know to prefer glucose (and so is called a glucophilic or 'glucose-loving' yeast). As fermentation proceeds the ratio of fructose to glucose increases. Therefore, fermented grape juice will contain more fructose than glucose as residual sugar. Fructose is the sweetest hexose sugar, approximately twice as sweet as glucose, and thus the wine gets an undesirable fructose sweetness, unbalanced by the sweetness of glucose.
So here is the starting point if you are doing a student experiment. Pose the Research Question: "How do the fermentation rates of glucose, fructose and sucrose compare?" Potentially it is a fantastic student experiment. Studies have found that glucose and fructose ferment at equal rates when they ferment separately; and when their concentrations are above 1%. However, when the glucose is below 1% it reacts faster than fructose. One delicious complication though is that when glucose and fructose are mixed (as in the case of fermenting sucrose; or in an artificial mixture) the glucose ferments faster than fructose. Glucose seems to inhibit fructose. Hmm - now that is tricky.
Your investigation could look at the fermentation rates of glucose and fructose separately - keeping everything the same except the independent variable of initial concentration. Note that 1% seems to be some sort of cut-off so examining concentrations either side of this would seem laudable. An then look at a mixture - or just look at sucrose as a natural mixture. What are you going to measure? The progress of the fermentation can be assessed by measuring the concentration of residual sugar or of the ethanol, or by the amount of CO2 produced. Sugar concentration can be measured using a Brix refractometer; or in the case of the two reducing sugars glucose and fructose, you can do a Fehling's titration; and there is a titration for ethanol. The density can also be used as an index. You could also monitor the reaction with a gas pressure sensor. I've used the Vernier sensor with a Texas Instruments CBL and that works well. There are lots of other gas sensors too.
A good start into the experimental glucose/fructose/sucrose fermentation is Leanie Mocke's 2013 Master of Science (Biochemistry) Thesis from Stellenbosch University, South Africa entitled Kinetic Modelling of Wine Fermentations: Why Does Yeast Prefer Glucose to Fructose? [available on-line].
6. Reducing sugars in wine 9.Carbon dioxide by titration 11. Polyphenol Index or Permanganate Index 12. Tests for reducing sugars (British Nutrition Foundation) 13. Fermentation notes
Sulfites in wine - titrations
Bottles of wine usually show the words "contains sulfites". For example, the label at the left is of a Sirromet Vineyard Selection NV Sparkling Wine. Sulfite/sulphite is a general term for sulfur dioxide (SO2) where sulfur has the oxidation state +IV. Sulfites are widely used in winemaking as a preservative to protect wines from oxidation and microbial spoilage but some people are sensitive to them. A friend of mine has enjoyed white wine with sulfites for decades without a problem, but has become sensitive in his older age. This sensitivity can cause a reaction that range from mild to severe: a dry 'asthma' cough is the most common. Thus, the government requires labeling of any food or beverage with a sulfite concentration of more than 10 ppm.
The term 'sulfites' is an inclusive term for SO2 (sulfur dioxide), HSO3- (also called hydrogen sulphite or bisulfite) and SO32- (sulfite). These three species are pH dependent but in wine with a pH of 3-4 the first two species predominate. Sulfur dioxide is widely used in winemaking because of its antioxidant and antibacterial properties. But you need the right amount: too little and the wine goes "off", too much and it has a pungent taste and smell. Two titration methods for the analysis of sulfur dioxide can be downloaded here. They are the Rankine Aspiration and the Ripper Method (also included below).
7. Sulfur dioxide by Rankine Aspiration 8. Sulfur dioxide by Ripper Titration 15. Sulfur dioxide availability vs pH Table
Sulfites in white wine - a gravimetric method
After reading an article about analysing for sulfite in wine I spoke to German wine chemist Dr Tom Mortier from the Faculty of Health and Welfare, University Colleges Leuven-Limburg, Herestraat, Belgium. He has written about it and I thought it may make a good student experiment. Tom said that the most common method for the analysis of sulfites in wine is the Ripper method (see above), which utilizes a direct titration of the wine with iodine using a starch indicator. Tom suggested we try a new analytical technique he has been working on (see May 2015 Journal of Chem Ed download here).
You can precipitate the sulfite in wine by adding strontium chloride solution. However, the normal pH of wine is about 3-4 and at that pH the sulfite is in the HSO3- form which won't react with the Sr2+. But if you raise the pH to 8 it will react and form a strontium sulfite precipitate which can be fltered and weighed.
Tom suggests: Take 100mL of wine, add 2.5 M NaOH dropwise until its changes to a dark brown colour. Check its pH - it should be about 8. Add 10% strontium chloride solution and it will go white if sulfites are present. Filter and weigh the precipitate. Calculate the %sulfite using stoichiometric calculations. This method is a bit vague but that's for you to experiment with.
Lastly, what could your hypothesis be? You could look at sulfutes and temperature; sulfites and whether they drop if a wine sample is exposed to air. This would be a fabulous student experiment. I'm going to try it after the holidays.
Oxidation of wine to vinegar 1 - titratable acidity Once a bottle of wine is opened and the air gets in it starts to go off (even if it is re-stoppered). It may not be noticeable for a couple of days. Initially, this off-taste is just oxidation of some of the flavour components but then it starts to get more and more acidic as the ethanol is oxidized to ethanal (acetaldehyde) and then to ethanoic acid (acetic acid) by acetic acid bacteria (called Acetobacter). This may be okay if you want to make wine vinegar but not so good if you want the wine to be drinkable. The question is: what factors affect the ability of the bacteria to oxidize the ethanol in wine.
Natural oxidation: leaving a sample of wine open to the air for a couple of days may make it taste "off" but this may not be a change in acidity but merely the flavour components oxidizing. It will take a bit longer to have sufficient change in acidity to be picked up by an acid-base titration. I took about 100 mL of white wine and filled a small bottle to the brim and put the lid on. I left another 100 mL on the bench in an open beaker. A week later I titrated them both using 0.100 M NaOH and a 20 mL aliquot of the wine (by pipette) and phenolphthalein indicator. The sealed wine had a pH of 3.02 ND gave a titre of 17.80 mL. The wine left open to the air had a pH of 3.13 and gave a titre of 21.65 mL. So, a good student experiment can be made of this.
Acetobacter oxidation: You could also give the wine a kick along and instead of relying on the acetobacter floating around in the air, you could add some. This is called 'inoculating' your wine. A simple way is to get some organic vinegar from a supermarket or health food store that states on the label that it contains the "mother" - this means acetobacter. Then just and filter out the cloud (which is the suspended acetobacter) and use some of that in your wine. See photos below.
I didn't want to filter out the acetobacter so I just added 2.0 mL of the vinegar to a beaker-full of wine. This would change the pH and titres I know but I though I should try. I put one sample in a sealed air-free bottle and the other in a beaker on the bench. After a week I titrated them against the 0.100 M NaOH as mentioned above. The sealed wine gave a titre of 18.15 mL and the air-exposed wine gave a titre of 24.95 mL. So, this wine oxidized too. I didn't do the pHs as there was no point. Great for a student experiment .
Some independent variables worth considering:
Aeration: As well, you probably need to give the wine plenty of aeration. Acetic acid bacteria are aerobic microorganisms and thus will not grow in anaerobic (without air) conditions. So, it might be a good idea to keep it stirred, or having a large surface area, or bubbling air through it (from a fish tank aerator perhaps). You need to have several treatments (different surface areas, or different lengths of exposure to the air) so you can graph the results. I suggest 5 treatments with duplicates of each.
Independent variables. You may guess temperature, but what about acidity (from the natural tartaric acid) or preservatives (perhaps SO2), or even the amount of ethanol in the wine (spirits, such as Vodka, with 45% ethanol concentrations don't turn to acetic acid). It would seem that a good EEI could be developed from oxidizing inoculated wine (perhaps by a controlled aeration of wine samples with an aquarium pump ($10) and measuring the acidity after a given time (eg aerate each sample for 24 hrs at the same bubble rate). The independent variables (IV) could be one or more of:
1. Temperature The optimum growth temperature for Acetobacter is 25-30°C, with no growth observed at 40°C. Weak growth was observed even as low as 10°C, but none at 8°C. 2. Acidity or pH Perhaps add some tartaric acid to have a range of starting pHs (the optimum pH for the growth of acetic acid bacteria is 5.5-6.3, however, these bacteria can survive at the low pH values of between 3.0 and 4.0 found in wine). A pH of 3.3 and lower is inhibitory to most bacteria in wine, but not to acetic acid bacteria. Maybe try pHs of 2, 3, 4, 5, 6. 3. Ethanol concentration. Ethanol is a good carbon source for acetic acid bacteria, but is also inhibiting at concentrations that are too high. One report I saw said that in wine containing 5% ethanol, only 58% of the Acetobacter was active and that this was reduced to only 13% in wine containing 10% ethanol. At 15.5% it seems all Acetobacter activity is inhibited (stopped). You could take some wine and add ethanol to it as the independent variable. The problem is - where do you get ethanol from? Your school may not have a licence to buy 100% (absolute) ethanol, or even the 95% azeotropic mixture (with water) so you may have to distill your own from shop-bought wine (ask your teacher before you bring any to school). From the density of the distillate (use an SG bottle) you can calculate how much to add to some fresh wine. 4. Additives. Sulfur dioxide should prevent the growth of acetic acid bacteria in wine and is sometimes used commercially for this purpose. SO2 in wine consists of the free form (molecular SO2, bisufite ions HSO3-and sulfite ions SO32-) and a bonded form. At normal wine pH only about 5% of the free SO2 occurs in the molecular form (which is the most active anti-microbial form) and the other 95% as bisulfite and sulfite ions. Concentrations of up to 20 mg/L of free SO2 will kill the bacteria. The simplest thing to do is to use powdered sodium metabisulfite which is available from home-brew shops (and from many health food stores as a anti-bacterial bottle wash for bottling fruit and so on). Try 0 to 25 mg/L free SO2. I have attached a great paper "The occurrence, control and esoteric effect of acetic acid bacteria in winemaking" by W.J. Du Toit, and I. S. Pretorius from the Department of Viticulture and Oenology, Institute for Wine Biotechnology, Stellenbosch University, South Africa. It was published in Annals of Microbiology, 52, 155-179 (2002). Click here to download.
1. Temperature The optimum growth temperature for Acetobacter is 25-30°C, with no growth observed at 40°C. Weak growth was observed even as low as 10°C, but none at 8°C. 2. Acidity or pH Perhaps add some tartaric acid to have a range of starting pHs (the optimum pH for the growth of acetic acid bacteria is 5.5-6.3, however, these bacteria can survive at the low pH values of between 3.0 and 4.0 found in wine). A pH of 3.3 and lower is inhibitory to most bacteria in wine, but not to acetic acid bacteria. Maybe try pHs of 2, 3, 4, 5, 6. 3. Ethanol concentration. Ethanol is a good carbon source for acetic acid bacteria, but is also inhibiting at concentrations that are too high. One report I saw said that in wine containing 5% ethanol, only 58% of the Acetobacter was active and that this was reduced to only 13% in wine containing 10% ethanol. At 15.5% it seems all Acetobacter activity is inhibited (stopped).
You could take some wine and add ethanol to it as the independent variable. The problem is - where do you get ethanol from? Your school may not have a licence to buy 100% (absolute) ethanol, or even the 95% azeotropic mixture (with water) so you may have to distill your own from shop-bought wine (ask your teacher before you bring any to school). From the density of the distillate (use an SG bottle) you can calculate how much to add to some fresh wine. 4. Additives. Sulfur dioxide should prevent the growth of acetic acid bacteria in wine and is sometimes used commercially for this purpose. SO2 in wine consists of the free form (molecular SO2, bisufite ions HSO3-and sulfite ions SO32-) and a bonded form. At normal wine pH only about 5% of the free SO2 occurs in the molecular form (which is the most active anti-microbial form) and the other 95% as bisulfite and sulfite ions. Concentrations of up to 20 mg/L of free SO2 will kill the bacteria.
The simplest thing to do is to use powdered sodium metabisulfite which is available from home-brew shops (and from many health food stores as a anti-bacterial bottle wash for bottling fruit and so on). Try 0 to 25 mg/L free SO2. I have attached a great paper "The occurrence, control and esoteric effect of acetic acid bacteria in winemaking" by W.J. Du Toit, and I. S. Pretorius from the Department of Viticulture and Oenology, Institute for Wine Biotechnology, Stellenbosch University, South Africa. It was published in Annals of Microbiology, 52, 155-179 (2002). Click here to download.
The total acidity can be measured by titrating against sodium hydroxide. Look up a table to get the best indicator (weak acid, strong base). The photos below shows Jennifer and Rachel - Yr 11 Chem students from Moreton Bay College - turning wine into vinegar.
TITRATABLE ACIDITY - METHODS
There is one main way for measuring total acidity and that is with a titration against sodium hydroxide using a phenolphthalein indicator. There is a problem in measuring acidity is when you have red wine as the pigments disguise the indicator colour change. Chemistry teacher Gareth Whittaker from Mary MacKillop College, Brisbane, advises the use of a pH probe rather than an acid-base indicator; and titrating to an end-point of pH 8.2 (this is called a potentiometric titration to distinguish it from a direct titration which uses a colour-change indicator). The method he uses (O24A-FD) comes from the Association of Analytical Communities, AOAC. Click here to download a copy. A pH of 8.2 is the international standard for TA endpoints in wine.
10.Titratable acidity 14. Natural acids in fruits and vegetables Table
Oxidation of wine 2 - sulfites as inhibitors of oxidation
The previous section suggested that sulfites can prevent the growth of acetic acid bacteria and hence restrict oxidation. In this suggestion we look at the effect of different initial SO2 concentrations, (0, 125, 250 and 375 ppm) on the oxidation of ethanol, specifically with reference to acetaldehyde (ethanal) bonding. My thanks to Year 12 Chemistry student Codi Baker-Lahey from St Andrew's Anglican College, Sunshine Coast, Queensland, Australia for this suggestion and some of her results. It was done in 2016 under the supervision of her chemistry teacher Mrs Larsen. Codi wrote:
The oxidation of ethanol occurs in two phases. The first is the oxidisation of ethanol to ethanal. Here, if SO2 is present it binds to the ethanal and prevents the next stage from occurring. If there is no or limited SO2 then the next stage will occur - which involves further oxidation - causing the ethanal to turn into ethanoic (aceti)c acid.
The investigation supported the hypothesis that an increase in SO2 concentration would reduce oxidisation. Generally, apart from a very small number of anomalies, a higher SO2 concentration resulted in a lower titratable acidity and higher ethanol content. The most likely reason for this yielded relationship was that a higher SO2 concentration allowed more SO2 to bond to ethanal, preventing a larger amount of ethanol from oxidation to the next stage of ethanoic acid.
Four identical samples of mock wine were prepared. Mock wine was used to remove other possible bonding substances other than acetaldehyde. Sodium metabisulphite was added to change the amount of SO2. Ethanol was monitored through an alcoholmeter and titration. It was predicted that the ethanol in all four samples would decrease as the SO2 cannot prevent this stage from occurring. This hypothesis was confirmed.
Oxidation of Wine 3 - rate of reaction In the above EEI, it was suggested that the acidity of the wine be measured after a set time (one day, or one week etc). I said that you could have acidity as the dependent variable (DV, as measured by titration), and one of: temperature, pH, %ethanol or SO2 as the independent variable (IV). However, another approach is to use "time" as the independent variable. In this case you would set up an experiment where air was blown through a sample of wine and the acidity measured at regular intervals (eg every hour for six hours; or every day for 5 days).
You would then plot acidity on the y-axis and time elapsed on the x-axis. The rate of reaction would be the slope of the line at a particular time. Does the rate vary over the whole time period? Does the rate vary as the acidity increases (is there a relationship)? Perhaps you could compare red and white wine. Does the red anthocyanin in the red wine act as an antioxidant as some people believe? What a fabulous EEI, and you'd even have some wine vinegar for your fish and chips afterwards.
WINE CHEMISTRY TEACHING RESOURCES
1. Basic Wine Chemistry ppt 2. Effects of pH on wine ppt 3. Fermentation Variables ppt 4. Monitoring Fermentations + Variables ppt 5. pH and Acidity in Wine ppt 6. Phenolic Compounds and Tannins in Wine ppt 7. Oxidative reactions ppt 8. Small Scale Winemaking ppt 9. Small Scale Winemaking Part 2 10. Sulfite Chemistry in Wine ppt 11. Winemaking in the Classroom Part 1 ppt 12. Winemaking in the Classroom Part 2 ppt 13. Winemaking in the Classroom Part 3 ppt
NON-EXPERIMENT RESEARCH QUESTIONS
If you are looking for a couple of non-experimental research questions then here are some witten as thesis statements:
1. "The pH and titratable acidity of wine are two measures of the same thing". 2. "You can calculate pH of wine if you know its titratable acidity". 3. "A chemist can measure change in wine as the ethanol is oxidized to acetic acid". As the tannins produce hydrogen peroxide the colour changes and then the peroxide changes the ethanol to ethanal and then on to acetic acid. The colour will change, the smell will be different, the ethanol decreases, the titratable acidity will increase.. and so on.
WINE CHEMISTRY PROFESSIONAL DEVELOPMENT
For teachers who would like to have a basic introduction to wine chemistry or would like to have a refresher, the Queensland College of Wine Tourism at Stanthorpe offers professional development workshops that may be useful. No recommendations are made as it is up to teachers to check it out (but reports have been good). It is called the "Teachers' Wine Chemistry Professional Development". Teachers are provided with an overview of the wine production process and the chemical processes/analysis that are conducted at each stage of wine production with an emphasis on the importance of chemistry concepts to quality wine production. A great one is 'Winemaker for a Weekend' - a two-day hands-on program looking at the wine making process, interspersed with informative master classes and a food and wine tasting.
Ginger Beer - avoiding the headaches
Investigating the production of alcohol in wine can give you a few headaches - particularly if you think someone will drink your experiment. Dr Gary Turner, HoD Xavier Catholic College (Hervey Bay) suggests that for Chemistry experiments a good alternative is ginger beer using a 7-day fermentation recipe (see later). This may be also suitable if your school doesn't allow you to have alcoholic beverages on campus - even in an experiment.
Ginger beer is made traditionally by the yeast fermentation of a mix of sugar, water and ginger. It is rarely produced commercially but often home brewed. The beverage produced industrially is generally not brewed (fermented), but carbonated with pressurized carbon dioxide. It is really just a soft drink, sweetened with sugar or artificial sweeteners. However, there are some manufacturers who still brew it the old way: in Queensland, Bundaberg Brewery produces an excellent brewed ginger beer. It is cloudy and if you hold the bottle up to the light and you'll see it's full of ginger pieces. A good EEI would be to brew your own at home or in the lab using one of the many recipes available on the internet (but do NOT drink it; not for an EEI).
Gary suggests this for his Year 12 EEI: First: You will be following a 'standard' procedure for making a simple beer (e.g. two-day ginger-beer) to give you the background skills and chemistry involved in making a beer, and to explore the factors involved. This section of the work will also require you to define which factors you can reasonably test in a school-laboratory, and which variables in the production that you can vary. A copy of the EEI task sheet is available for download here.
"The students can try the variables of yeast, sugar, and temperature (by dividing batches into samples) and titrate for alcohol at intervals (many hands makes light-work). You need a fridge and an incubator to give you three temps (including room temp) for the temp-as-variable". His method is one of many that can be easily obtained from the internet:
You need to create what is called a Ginger Beer Plant. Put 15g of general purpose dried yeast into a large jar or bowl, add 300mL water, 2 teaspoons ground ginger and 2 teaspoons sugar. Cover with a sheet of cling film and secure with a rubber band. Each day, for seven days, add 1 teaspoon of ginger and 1 teaspoon of sugar to the mixture in the jar. Now strain the mixture through a piece of fine muslin and add the juice of two lemons to the liquid. Add 50g or sugar to the liquid and make up to 4.5 litres with cold water, stirring to dissolve the sugar. Bottle into a plastic bottle Keep for 7-10 days when the ginger beer is sparkling and ready for drinking.
You can keep the sediment that you have left after straining the ginger beer plant. Divide into two jars and give 1 plant away to a friend with the instructions. To the sediment add 300 mL water, 2 teaspoons sugar and 2 teaspoons ginger and carry on as before.
Alcohol is produced but its concentration is likely to be under 1%. Also, most fermented soft drinks are acidified to inhibit bacterial growth. Does this also inhibit the yeast? You could investigate the effect of pH on the rate of fermentation using lemon juice or better - citric acid. The juice of 1 lemon contains about 12 g citic acid. Be warned - you should not be drinking the ginger beer unless you have approval from your teacher (and this is unlikely). Drinking stuff made in a laboratory with no hygiene controls is DEFINITELY NOT PERMITTED.
Lastly, what sort of yeast is best? Chemistry teacher Torsten Pluschke of Atherton State High School (North Queensland) said that you could buy genuine ginger beer plant (a SCOBY – symbiotic colony of bacteria and yeast, rather than just baker/brewer's yeast) online. He said that it must be looked after, and consumed in a shorter time frame than baker/brewer or wild yeasts. One online site (no recommendations given though) is The Ginger Beer Plant who can provide a 50 g sachet of the SCOBY for about $20 including postage to Australia. I asked at my local brew shop and the guy said none was available in Australia and it had to be bought online.
However, what yeast do you think Bundaberg Brewed Drinks use for their Brewed Ginger Beer? Here's what Richard Cowdroy-Ling, General Manager Business Technologies, told me about their product:
When you pick fruit of a tree or vine it remains alive even though it is separated from its parent. They can't get water or nutrients from the parent so they have to use their own stored chemicals to continue. Bananas are like this. Mature green bananas are full of starch, but when you pick them they begin to ripen: their average starch content just before ripening reaches 25% and drops over a few days of ripening to less than 1%. But little is known about the mechanism involved.
It seems that the hormone ethylene triggers the production of enzymes. For example, the alpha (1 to 4) bonds of starch may be hydrolyzed by amylases (and glucosidases) or broken by starch phosphorylases. It has been observed that sucrose starts to accumulate first, before glucose and fructose, and parallel to starch disappearance. See N. Terra et al, 1983. "Starch-Sugar Transformation during banana ripening". Journal of Food Science, V48(4) p1097-1100.
This suggests a great EEI (which could be equally as good for Biology). The question can be asked "What conditions affect the kinetics of banana ripening?" The obvious one is temperature, but an intriguing one is the presence of ethylene (ethene). This gas is produced as bananas begin to ripen so it would be instructive to compare bananas ripening in a plastic bag where the ethylene is trapped in with the fruit, versus ripening in a breeze where the ethylene is blown away. I've used a little fan out of a computer. That's all you need.
What would you measure as an index of ripening? Some books suggest iodine but that is too inaccurate for a Chemistry EEI. I suggest measuring the concentration of glucose. There are a couple of methods of measuring this but the two main ones would have to be: (a) using glucose test strips either by comparison against colour standards, or by use of a glucometer as used in diabetes monitoring; (b) by titration using either using Benedict's solution, or an iodine/thiosulfate titration (download).
Antioxidants in food and the effect of temperature
Antioxidants are a wide variety of compounds found in fruit and vegetables that are beneficial for health because they destroy free radicals and prevent tissue damage, especially in blood vessels. They are often the colorful pigments in these foods. For example, the anti-oxidant beta-carotene provides the orange hue in carrots, lycopene gives tomatoes their red appearance and anthocyanins make blueberries and certain grapes look dark purple.
The temperatures used for cooking in most household kitchens are enough to destroy particularly heat-sensitive antioxidants such as vitamin C, but the antioxidants in some foods actually become more potent with heat. For example, when tomatoes are cooked for 30 minutes at 88°C, they lose almost 30% of their Vitamin C, but 35% more of the anti-oxidant lycopene becomes available. Beta-carotene levels in carrots also increase with moderate heat. The reason seems to be that the heat breaks down the plants' thick cell walls and makes the nutrient available. [See below for reference].
This suggests a great EEI. The problem is - how can we measure antioxidant concentration in the laboratory. The Briggs-Rauscher reaction, an oscillating chemical reaction that exhibits a vivid colour change from colourless to amber to a sudden dark blue, can be used to determine relative antioxidant concentration in foods (and wine). The reaction is very complex and involves both iodide ions and iodine molecules. It is an "oscillating" reaction that goes from blue to colourless to yellow to blue in a period of time that depends on the relative concentrations of these species.
WARNING: Before you get too carried away with this experiment, you should be aware that one of the chemicals involved is malonic acid. This is not too hard to get but you need to plan ahead. I bought some through Labtek (Australia) but it took almost 4 weeks. The one I got was Ajax 'Unilab' brand, code 305-100G, $63.80 for a 100 g bottle. Griffith University would have helped as part of their outreach program to schools out but they would have to order it in too. I know of three girls at Palm Beach Currumbin State High School (hello to Jissa, Momoka and Niki) on the Gold Coast, Queensland, who started to do this EEI but ran into time constraints. They told me that they found a website "that may supply future students with a kit, inclusive of malonic acid, that will allow for them to do the BR reaction: http://www.teachersource.com/product/fascinating-oscillating-reaction-kit/chemistry."
Using this reaction, it is possible to determine the level of antioxidants present in everyday foods and drinks by starting a timer when the reaction mixture is blue and noting how long it takes for the reaction mixture to go through one full cycle (stopping the stopwatch when the mixture is blue again). The addition of antioxidants increases the time taken for this reaction. The longer the time taken for the reaction cycle, the more antioxidants the food contains. A terrific resource which includes a detailed method is available at Learning and Teaching Scotland.
Factors affecting the viscosity of cellulose gum
Carboxymethylcellulose (CMC) is a modified cellulose gum that can be used as a food thickener (Code E461). It produces a clear, slightly gummy, solution in water. It is used to thicken dry mix beverages like soups, as well as syrups and ice cream. In the wine industry it has recently been approved as an additive to keep wine clear. CMC acts upon the face of a growing crystal, restricting further growth while ensuring that nothing is visible to the naked eye. Metal ions with different charges significantly affect the viscosity of aqueous sodium carboxylmethylcellulose solution. And herein like the essence of a great EEI.
You could take an aqueous sodium CMC solution and add various metal ions to it. You could try metal salts such as LiCl, NaCl, KCl, CaCl2, AlCl3 and make aqueous 0.50% (w/v) solutions in the range 0.00-0.10 mol/kg. To measure viscosity you could use a tube (say 2 cm diameter) and about 100 cm long (or even a 100 mL measuring cylinder). Put marks as far apart as possible, maybe 50cm apart (or at the 80 and 20 mL marks on the measuring cylinder). Fill it with the solutions and drop small (plastic) balls and time between the marks. You want balls that are not too dense but will not float (about 1.020 g/cm3 seems okay). Better note the temperature.
The viscosity of the sample was then calculated from the following (Poiseuille) equation η/ηo = ρt/ρoto where ηo (viscosity) is in mPa s, ρo (density) is in g cm-3, to (flow time) is in s, ρ is in g cm-3. The standard values of viscosity (ηo) and density (ρo) for water at 25 °C can be taken from the literature, while the densities of samples can be measured by whatever method you like.
The graph shows that metal ions have significant effects on the viscosity of aqueous 0.50%w/v CMC solutions and its extent is particularly dependent on metal ion charges. NaCl and LiCl and the same as KCl. The cation size effects of 1+ metal ions such as Li+, Na+, and K+ on viscosity are all very similar. The viscosity of the aqueous CMC solutions gradually decreases with the increase of alkali metal chloride concentration. On the other hand, the effect of calcium ion with 2+ charge is more dramatic. The CMC viscosity decreases much more quickly with the increase of CaCl2 concentration. However, in the case of 3+ cations such as Al3+ you will get a real surprise.
My thanks to Seng Set and Masakazu Kita, Department of Chemistry, Okayama University, Japan, and David Ford Department of Chemistry, Royal University of Phnom Penh, Vietnam. Refer J. Chem. Educ. 2015, 92, 946-949.
Nitrification in Soils - a great titration prac. A great EEI concerns the process of nitrification by soil bacteria. It would be suitable for either Chemistry or Biology. It is also broadly related to wastewater treatment and maintenance of fish aquariums.
Nitrogen is one of the most essential nutrients for plants and is most frequently the limiting factor in crop productivity. The vast majority of the total nitrogen in soil (>98%) is in organic matter, which can't directly be used by plants. It must be converted to inorganic forms as either ammonium (NH4+) or nitrate (NO3-). This conversion occurs via a biological process called nitrification involving soil microorganisms.
Nitrification in nature is a two-step oxidation process of ammonium ion or ammonia to nitrate ion catalyzed by two types of bacteria. The first reaction is oxidation of ammonium to nitrite by ammonium oxidizing bacteria (AOB) represented by the Nitrosomonas species. The second reaction is oxidation of nitrite (NO2-) to nitrate by nitrate-oxidizing bacteria (NOB), represented by the Nitrobacter species.
The progress of nitrification can be investigated using a fairly common titration procedure. It involves making a filtered solution of the soil, boiling off free ammonia, adding an excess of sodium hydroxide solution (which reacts with the ammonium ion) and back-titrating the excess hydroxide with standardized hydrochloric acid. The method can be easily found on the internet and may even be in your textbook under a heading of "analyzing fertilizer". The photos below are courtesy of Yr 12 student Jamie from Our Lady's College, Annerley, Brisbane.
A good mixed source of nitrifying bacteria and ammonium ions is potting mix. It usually contains peat moss, sand, and other organic material such as wood chips. It would have reasonable amounts of ammonium ions present. Your EEI could be to measure the ammonium ion concentration after a period of time (say a week) under different conditions that may affect the bacteria (temperature, oxygen availability, pH, salinity, light).
Temperature: nitrifiying bacteria are supposed to be at their optimum from 25°C-30°C. Aquarium operators have a rule of thumb that says: 50% activity at 20°C, 25% at 10°C, 0% at 4°C and are death at 0°C. When heated, their activity decreases after 30°C and they die by 50°C. That suggests a great investigation. Oxygen: the bacteria need oxygen to produce energy to live (respiration). If you cut off their oxygen supply their activity decreases. Farmers know that in waterlogged soils the bacteria are less productive. That suggests waterlogging your potting mix samples to prevent the bacteria getting oxygen and comparing them to the control. It would be good to know how much water is present in the potting mix and you can do this by taking a weighed sample and drying it in the oven at 100°C (gravimetric analysis). Because the hydroxide is in the flask, the end point will be when the pink just disappears. The flasks at the end point. Moisture: nitrifying bacteria are most active in a soil that is moist but not saturated (see above). Is there an optimum amount of moisture? If completely dry they go into hibernation - but what is optimum? pH: there is an optimum for the first stage bacteria (nitrosomanas) of 7.8 to 8.0, and for stage two (nitrobacter) it is a little bit less. This investigation would require careful design. Potting mix is usually pH 5-6.5 so you'll have to add some alkali to increase the pH. This will affect the acid/base titration. You'll need to think about a controlled sample without potting mix but with the added base. As a matter of interest, as the ocean becomes enriched in anthropogenic (human activity) CO2 the resulting decrease in pH could lead to decreasing rates of nitrification. That's another context for this EEI. Salinity: the optimum is said to be zero to 0.6%. Light: the nitrifying bacteria are supposed to be sensitive to blue/UV light. But how far into the soil would light penetrate (a good design consideration).
Temperature: nitrifiying bacteria are supposed to be at their optimum from 25°C-30°C. Aquarium operators have a rule of thumb that says: 50% activity at 20°C, 25% at 10°C, 0% at 4°C and are death at 0°C. When heated, their activity decreases after 30°C and they die by 50°C. That suggests a great investigation.
Oxygen: the bacteria need oxygen to produce energy to live (respiration). If you cut off their oxygen supply their activity decreases. Farmers know that in waterlogged soils the bacteria are less productive. That suggests waterlogging your potting mix samples to prevent the bacteria getting oxygen and comparing them to the control. It would be good to know how much water is present in the potting mix and you can do this by taking a weighed sample and drying it in the oven at 100°C (gravimetric analysis).
Moisture: nitrifying bacteria are most active in a soil that is moist but not saturated (see above). Is there an optimum amount of moisture? If completely dry they go into hibernation - but what is optimum?
pH: there is an optimum for the first stage bacteria (nitrosomanas) of 7.8 to 8.0, and for stage two (nitrobacter) it is a little bit less. This investigation would require careful design. Potting mix is usually pH 5-6.5 so you'll have to add some alkali to increase the pH. This will affect the acid/base titration. You'll need to think about a controlled sample without potting mix but with the added base. As a matter of interest, as the ocean becomes enriched in anthropogenic (human activity) CO2 the resulting decrease in pH could lead to decreasing rates of nitrification. That's another context for this EEI.
Salinity: the optimum is said to be zero to 0.6%.
Light: the nitrifying bacteria are supposed to be sensitive to blue/UV light. But how far into the soil would light penetrate (a good design consideration).
My thanks to Chemistry colleague at Our Lady's College, Annerley, Brisbane, Kayleen Solomon and Yr 12 student Jamie Nguyen for providing ideas and photos for this EEI suggestion. A copy of Jamie's method can be downloaded here.
Nitrification in Soils - spectrographic determination
In the above suggestion, a back titration was used to determine the ammonium concentration. If you have access to a spectrophotometer and the reagents required you could determine nitrate ion spectrophophotometrically.
Physically, nitrite is a colorless and odorless ion. However, there are spectrophotometric methods for its determination. One example is to react it with an acidified sulfanilamide and N-(1-naphthyl)ethylenediamine dihydrochloride to form an intensely coloured dye having maximum absorption at 540 nm. There are lots of other methods. Often you can get them in the form of test kits where you compare the colour to a test strip. Before you get started check that you can get the reagents.
Stability of Vitamin C in solution - analysed by titration. Vitamin C is sensitive to heat and oxygen and the degree of sensitivity depends on the pH of the solution. In food it can be partly or completely destroyed by long storage or overcooking. By refrigeration the loss of Vitamin C in food can be substantially diminished. An interesting EEI would be to see how some of these factors really affect a Vitamin C solution. You could start with some fresh fruit juice (eg apple, orange) or you could simulate fruit juice by making up an appropriate solution with added citric acid, some citrates, glucose/fructose and so on. Should you measure the concentration of the ascorbic acid with time (eg daily) or just measure after a week or two weeks? What will you control? What will your independent variable be: sugar concentration, [H+], light, oxygen, temperature? If you intend to measure the concentration as a function of time elapsed you should read my caution below.
The possibilities are endless but you'd need to back up your hypothesis with some justification from the literature. Your hypothesis could be in the following form: "That the concentration of ascorbic acid in solution decreases faster with time as the [manipulated variable] is increased/decreased".
Here are some data to get you started. In an examination of some Vitamin C products for the pharmaceutical industry Uprety and Revis (1964) reported that the factors below were important (but not necessarily linear in their response to the manipulated variable). There were a lot of anolomous data in their results.
Temperature: after 30 days 20% of the ascorbic acid was lost at 37°C, whereas 46% was lost at 55°C. pH and acidity: after 30 days 33% of the ascorbic acid was lost at pH 2.5 and 35% lost at pH 6.5, thus low pH seems to stop the loss of ascorbic acid. However, there were lots of anomalies and in some samples the losses went down as the pH increased. Also note that when fruit is cut, the enzyme polyphenol oxidase is released from the cells and reacts with the oxygen in the air causing the fruit to deteriorate. When you decrease the pH by adding citric acid you tend to stop the polyphenol oxidase working as its optimum range is from pH 5 to 7. In fact, below a pH level of 3.0, the enzyme is inactivated. In the Uprety and Revis article it was found that citric acid was protected ascorbic acid well. An apple juice sample lost 98.6% of its ascorbic acid after 60 days, but when 0.2% citric acid was added it lost just 59% (they didn't say the pHs. The pH of lemon juice is in the 2.0 range, making it very effective against browning. Now there's a great EEI. Antioxidants: Citric acid can be considered to be an antioxidant but mainly for its low pH rather than anything special about the citric acid molecule (other fruit acids would work if their pH was low enough). However, it is also known that EDTA (ethylene diamine tetraacetic acid) protects juices in certain cases. It is believed that the EDTA binds any copper ions present. A 0.01% solution seems to work with apple juice. Sodium chloride also seems to work - a 0.9% solution is believed to counter the oxidation of ascorbic acid. Natural juice vs water: A sample of lime juice at pH 3.5 for 30 days at 37°C lost 76% of its ascorbic acid, whereas distilled water at the same pH and temperature lost only 50% in the same time. Now that discounts the pH argument. There must be something else that goes on too. Different juices: some juices have substances which help destroy the ascorbic acid. For example, apple juice held at 37°C, pH 3.5 for 60 days lost 98.6% of its ascorbic acid, whereas pineapple juice under the identical conditions lost just 71.1%. This is attributed to the presence of polyphenol-oxidase in the apple juice which catalyses the oxidation of ascorbic acid. Head Space: the bigger the headspace in a sealed container the greater the loss of ascorbic acid. The more access the juice has to oxygen the greater the degredation. Bubbling air through a sample would speed things up you would think. Colour change: as ascorbic acid breaks down the juices got darker in colour. This is a good index of ascorbic acid concentration if you have a colorimeter. See my smartphone colorimeter suggestion later on this webpage. There's a free app from your iPhone or Android that will turn them into a colorimeter. Works well.
Temperature: after 30 days 20% of the ascorbic acid was lost at 37°C, whereas 46% was lost at 55°C.
pH and acidity: after 30 days 33% of the ascorbic acid was lost at pH 2.5 and 35% lost at pH 6.5, thus low pH seems to stop the loss of ascorbic acid. However, there were lots of anomalies and in some samples the losses went down as the pH increased. Also note that when fruit is cut, the enzyme polyphenol oxidase is released from the cells and reacts with the oxygen in the air causing the fruit to deteriorate. When you decrease the pH by adding citric acid you tend to stop the polyphenol oxidase working as its optimum range is from pH 5 to 7. In fact, below a pH level of 3.0, the enzyme is inactivated. In the Uprety and Revis article it was found that citric acid was protected ascorbic acid well. An apple juice sample lost 98.6% of its ascorbic acid after 60 days, but when 0.2% citric acid was added it lost just 59% (they didn't say the pHs. The pH of lemon juice is in the 2.0 range, making it very effective against browning. Now there's a great EEI.
Antioxidants: Citric acid can be considered to be an antioxidant but mainly for its low pH rather than anything special about the citric acid molecule (other fruit acids would work if their pH was low enough). However, it is also known that EDTA (ethylene diamine tetraacetic acid) protects juices in certain cases. It is believed that the EDTA binds any copper ions present. A 0.01% solution seems to work with apple juice. Sodium chloride also seems to work - a 0.9% solution is believed to counter the oxidation of ascorbic acid.
Natural juice vs water: A sample of lime juice at pH 3.5 for 30 days at 37°C lost 76% of its ascorbic acid, whereas distilled water at the same pH and temperature lost only 50% in the same time. Now that discounts the pH argument. There must be something else that goes on too.
Different juices: some juices have substances which help destroy the ascorbic acid. For example, apple juice held at 37°C, pH 3.5 for 60 days lost 98.6% of its ascorbic acid, whereas pineapple juice under the identical conditions lost just 71.1%. This is attributed to the presence of polyphenol-oxidase in the apple juice which catalyses the oxidation of ascorbic acid.
Head Space: the bigger the headspace in a sealed container the greater the loss of ascorbic acid. The more access the juice has to oxygen the greater the degredation. Bubbling air through a sample would speed things up you would think.
Colour change: as ascorbic acid breaks down the juices got darker in colour. This is a good index of ascorbic acid concentration if you have a colorimeter. See my smartphone colorimeter suggestion later on this webpage. There's a free app from your iPhone or Android that will turn them into a colorimeter. Works well.
Titrations In a high school lab, the easiest way to measure ascorbic acid concentration is by titration. There are two common methods, both of which work well. The first is by DCPIP titration. DCPIP is 2,6-dichlorophenolindophenol and reacts with ascorbic acid in a 1:1 ratio. It is a blue dye that produces a nice pink to colourless end point but is quite expensive. A 1.0 g bottle costs about $45 (but you only need about 0.2g per litre) - available from Rowe Scientific, Brisbane Ph 3376 9411, email roweqld@rowe.com.au.
A DCPIP method that has been trialled extensively features a phosphoric/acetic acid extracting solution. I have adapted this method and added sample calculations. It can be downloaded by clicking the link: Vitamin C DCPIP Method. The one caution with DCPIP and the cause of so much misery amongst students is that DCPIP is not easy to dissolve; you need to leave it overnight and then decant and filter it the next day.
THE COLOUR OF DCPIP IN ACID
The blue DCPIP solution goes pink when it mixes with acid - any acid.This is its acid colour. It does this even when no ascorbic acid is present. Students get confused because they think the pink is the colour DCPIP changes to when it reacts with ascorbic acid. It is not. When it reacts with ascorbic acid the pink DCPIP goes colourless. When all of the ascorbic acid is used up (at the end point) there is no ascorbic acid left to make it go colourless so it goes pink (permanently).
STANDARDIZING DCPIP WITH STANDARD ASCORBIC ACID SOLUTION
TITRATING ORANGE JUICE WITH DCPIP
IODINE TITRATION OF ASCORBIC ACID IN FRUIT JUICE
The iodate ion IO3- reacts with the iodide ion I- in acidic H+ solution to form iodine I2 and water H2O.
2 IO3- +10 I- + 12 H+ → 6 I2 + 6 H2O
During the titration the iodine reacts with ascorbic acid C6H8O6 in your sample to form dehydroascorbic acid C6H6O6, iodide ions I- and hydrogen ions H+
6 I2 + 6 C6H8O6 → 6 C6H6O6 +12 I- + 12 H+
When all of the ascorbic acid is used up by the iodine at the end point, the excess iodine turns the starch blue.
The overall reaction can be seen by the addition of the above reactions and the coefficients simplified:
IO3- +5 I- + 3 C6H8O6 → 6 I- + 3 C6H6O6 + 3 H2O
The stoichiometric ratio is:
1 IO3- + 3 C6H8O6 or n (IO3-)/1 = n (C6H6O6)/3
C iodate x V iodate = m vitc/ M vitc/3
25 mL orange juice and the first drops of the iodine solution
At the end point.
For a copy of this method with sample calculations click:Vitamin C Iodine titration.
My thanks to Tara Robinson - Yr 12 Chem at MBC - for the photos.
TITRATION OF SODA WATER
Rate of carbon dioxide loss in soda water as it goes flat. A great modification.
Here's a suggestion that could make a great modification to the mandatory practical: Acid-base titration to calculate the concentration of a solution with reference to a standard solution. [QCAA Chemistry 2019 v1.2 General Senior Syllabus, p 42]. The syllabus makes it clear that you should be aware that carbonated water is a weak acid (page 41):
Soft drink ('soda' or 'pop' in the USA) goes flat when you leave it out for a day or so. When the lid is on there is an equilibrium between the dissolved carbon dioxide in the liquid and the CO2 gas in the headspace. When the lid is opened the pressure is relieved and the equilibrium is disturbed. According to Le Chatelier's Principle the system tends to oppose the removal of the gas from the headspace so more and more carbon dioxide comes out of solution to replace it. As the gas can never build up sufficient pressure above the surface of the liquid, the gas keeps coming out of the solution until it is 'flat'. But we can measure the [CO2(aq)] in the drink by titration as the CO2 forms the weak acid H2CO3 (or in its dissociated form HCO3- (aq) + H+(aq)), and this can be titrated against a standard solution of the strong base NaOH. In soda water the CO2 is present in its aqueous form CO2 (aq). You can't just measure the acidity of the soda water at one point in time like you might for the mandatory experiment. For a good student experiment you need to show a relationship that can be plotted, such as how the acidity changes as a function of time.
To avoid the complication of residual acidity caused by the presence of citric acid (or phosphoric acid in the case of cola drinks), it is simpler to use plain soda water - which has no added acid or acidic fruit syrups.
The reactions for the titration can be represented simply as: 1. CO2 (aq) reacting with water to form a weak acid: CO2 (aq) + H2O (l) → HCO3- (aq) + H+(aq) 2. Titration of the weak acid with aqueous NaOH: HCO3- (aq) + H+(aq) + 2 NaOH → Na2CO3 + H2O 3. Summary titration reaction: CO2 (aq)+ 2 NaOH (aq)→ Na2CO3 (aq)+ H2O (l)
The stoichiometric relationship for the titration can be represented as:
HINT 1. Check the label and make sure the drink is just water and carbon dioxide. It will say 'carbonated water'. Other carbonated drinks such as Club Soda and 'sparkling mineral water' contain added or dissolved minerals such as potassium bicarbonate, sodium bicarbonate, sodium citrate, or potassium sulfate. Use 'soda water' if you can to avoid the presence of other substances. If you can't find soda water, use sparkling mineral water.
HINT 2. The titre for flat soda water should be (almost) zero but if it is not then you have some further checking to do. Check the label again to see that it is 'carbonated water' and not 'carbonated mineral water'. If it is just carbonated water then it is likely you have contamination of the solutions. Perhaps the flasks had some residual HCl in them from an earlier standardisation. Rinse everything again and redo the titrations.
HINT 3. To meet the syllabus criterion of collecting 'sufficient' data the 5 x 3 approach is genarally recommended. That is: 5 variations of the independent variable (time elapsed) and 3 repetitions at each variation. This is appropriate when the relationship is linear or almost linear. However, the rate of carbon dioxide loss from the drink is fast initially and then tapers off as time elapses. The acidity thus decreases in an exponential or a logarithmic manner. When this happens, the change at the start is fast and so the graph curves sharply. It is therefore recommended that you do more than 5 variations (maybe 7 or more). So do more in the first few hours on the first day and one more the next day. At 24 hours the sample should be almost flat but is worth including. eg 10, 20, 40, 80, 120, 240 minutes, and one the next day, and a final one the day after that. Check my setout below. I've used clear plastic party cups from Spotlight as most schools won't have enough flasks or beakers when there is high demand during prac work. Mine were Party Creator Plastic Cups Clear 450 mL 20 cups for $1.50.
HINT 4. I suggest you do three replicate titrations for each of the 7 (or more) trials. For replicates, you could have three separate 25.00 g samples for each time interval. For this approach I suggest you weigh out 25.00 grams of the soda water into each of 21 containers (beakers or party cups) at the start. You can't pipette up 25 mL aliquots of the soda water as the liquid bubbles in the pipette and goes all over the place. That's why I said to weigh out your samples. The syllabus criteria for the number of samples to use is that they are 'sufficient'. This is defined as 'suitable for the purpose', or as QCAA advises (Online Webinar, 13 June 2022) it needs to be sufficient to answer the research question and therefore can't be assigned an arbitrary amount. Thus, a 5 x 3 approach is quite sufficient for a linear relationship, but 7-8 x 3 is better for a non-linear one. In particular, you need closer increments for the independent variable (on the x-axis) when the change in the dependent variable (on the y-axis) is great.
HINT 5. At each time interval titrate three of the samples. That gives you three equivalent replicates and the variation between the replicate titres is likely to be small. Ideally, you would like the replicates to be 'concordant', that is, within 1 drop of each other (that is, +/- 0.05 mL). Aim for that but expect the variation to be higher (maybe +/- 0.2 mL) as it hard to control bubbly drinks that constantly off-gas (lose gas during the titration). The syllabus criteria makes no mention that they need be 'concordant' as this is almost impossible for carbonic acid that is continually off-gassing. Averaging the three titres and determining the uncertainty would enable you to make a 'thorough and appropriate identification of the uncertainty' [QCAA Chemistry 2019 v1.2 General Senior Syllabus, p 54].
HINT 6. The end point for a weak acid/strong base titration can be determined using phenolphthalein indicator. The end point is marked when the faint pink colour lasts for 30 seconds. My video below shows this:
HINT 7. Your titres are likely to be less than 10 mL so you may like to use 0.05 M NaOH instead of 0.1 M NaOH. It's up to you. Alternatively, use 50 mL (50 g) aliquots. I do it with 25 g aliquots and get titres in the range 2-6 mL which gives me reasonable uncertainty for this purpose.
HINT 8. Keep the lid on the 0.1 M NaOH solution unless you are filling the burette. NaOH solution will absorb CO2 from the atmosphere and react to form sodium carbonate. This reduces the concentration of the NaOH solution and will add a systematic error to your titres.
HINT 9. You need to decarbonate some soft drink to use as a control. Heating it to say 80o C is the simplest way to achieve this. No need to boil it though. It should have a titre of 0-0.05 mL (1 drop). You can subtract this from each titre if you like but is not essential for developing a relationship. Mention it if you like.
RESULTS. Here are some of my results. I've just shown the average titres for 25 g aliquots of the soda water titrated against 0.100 M NaOH. It was impossible to get concordant titres (three within 0.05 mL of each other) so I've just used averages as explained previously.
t (min)
10
25
45
75
135
210
380
895
2400
degassed
Titre (mL)
5.85
4.85
4.52
4.13
3.58
2.98
2.47
1.60
0.80
0.10
CALCULATIONS FOR CARBON DIOXIDE Volume of soda water aliquot (Vacid) = 25.0 mL Concentration of standard sodium hydroxide solution (CNaOH) = 0.100 M Reaction: CO2 + 2 NaOH → Na2CO3 + H2O CacidVacid = CNaOHVNaOH/2 Cacid= CNaOHVNaOH/(2 x Vacid) Sample calculation for the titration of 25.0 mL of soda water at 10.0 minutes. Titre (VNaOH) = 5.85 mL Cacid= CNaOHVNaOH/(2 x Vacid) Cacid= 0.100 x 5.85/(2 x 25.0) Cacid = 0.0117 M
FURTHER CALCULATIONS. In industry, the concentration of CO2 is usually expressed as a percentage (%CO2w/v). To convert from molarity (mol L-1) you just multiply by the molar mass of CO2 (44 g mol-1) to give g/L, and then divide by 10 to give g/100 mL ( = %CO2w/v). For example: Cacid = 0.0117 M Cacid = 0.0117 x 44 = 0.5148 g/L Cacid =0.5148/10 = 0.05148 g/100 mL = 0.05148 %(w/v)
You graph will have different values but the shape should be similar. Note the custom error bars. In a chemistry 'student experiment' you may not need to include error bars. Check with your teacher. If you are not sure of how to calculate uncertainty and display error bars have a look at my YouTube video: https://youtu.be/mVS7YWENShs
THEORY BEHIND RATE OF DECARBONATION POINT 1. With the lid on, there is an equilibrium between carbon dioxide gas in the headspace and the dissolved carbon dioxide in the water. Henry's Law gives a relationship between the pressure of the CO2 gas in the headspace and the amount of CO2 gas dissolved in the water. Henry's Law is P = kHC. Here's a sample question and calculation to show how it applies. Question: Calculate the mass of CO2 in 1.00 L of soda water under a headspace pressure of 3.50 atmospheres (atm). Note: Henry's Law constant kH = 29.76 atm mol L-1. Solution: Henry's Law (P = kH C) can be rearranged to give C = P/kH C = (2.50 atm)/(29.76 atm mol L-1) = 0.084 mol L-1. n = m/M, thus m = nM m = 0.084 mol x 44 g mol-1 = 3.70 g (to 3 significant figures).
Note: This is the mass CO2 in 1 L of soda water. We could further calculate the concentration of CO2 in the soda water in g/L, thus: C = m/V = 3.70 g /1.0 L = 3.70 g L-1. This is equal to 0.370 g/100 mL, which is the same as 0.370 % (w/v).
POINT 2. You can use Henry's Law to work out the theoretical concentration of CO2 in water when exposed to just normal atmospheric pressure (1 atm, or 101.3 kPa). The current atmospheric concentration of CO2 is 360 micro-atmospheres (360 μ atm). It is rising because of human action (or inaction) but let's just use the accepted value for the moment. Question: Calculate the mass of CO2 in 1.00 L of water at a pressure of 1.00 atmosphere (1.00 atm). Solution: Henry's Law: C = P/kH = (360 x 10-6atm)/(29.76 atm mol L-1) = 1.21 x 10-5 mol L-1 n = m/M, thus m = nM = 1.21 x 10-5 mol x 44 g mol-1 = 5.32x 10-4 g in 1 litre (3 sf). Note: This is written as: C = 5.32x 10-4 g L-1.
POINT 3. For water exposed to the atmosphere (atmospheric pressure), the titre with 0.100 M NaOH can be calculated. The mole ratios in the titration reaction are: CNaOHVNaOH/2 = CCO2VCO2 VNaOH = 2 x (CCO2VCO2)/CNaOH= (2 x 1.21 x 10-5 x 20.00)/0.100 = 0.0024 mL. This is about 1/20th of a drop of base (0.05 mL) of 0.100 M NaOH, so the titre should be zero.
POINT 4. When CO2 dissolves in water, the following sequence of reactions takes place: CO2 (g) → CO2 (aq) CO2 (aq) + H2O (l) → H2CO3 (aq) H2CO3 (aq) + H2O (l) → HCO3- (aq) + H3O+(aq) HCO3- (aq) + H2O (l) → CO32- (aq) + H3O+(aq) Or, in summary: CO2 (g) + H2O (l) → CO32- (aq) + 2H+(aq) When the pressure is released, the equilibrium shifts towards the left (reactants). Thus, the CO2 (g) comes out of solution in the form of bubbles.
POINT 5. The rate of decarbonation depends on four processes: 1. The speed of formation of nucleation sites in the soda water. These are places in the body of the liquid where bubbles can form. They may be scratches or imperfections in the surface of the container, impurities in the liquid, or even other bubbles. 2. The rates of the reverse reactions. That is, the rate at which the carbonate ion (CO32-) and hydrogen carbonate ion (HCO3-) revert to carbonic acid (H2CO3) and thence to dissolved CO2 (aq) and finally gaseous CO2. This is controlled by the activation energies of the reverse reactions and any rate determining steps. 3. The speed of formation of the bubbles at the nucleation sites. This is controlled by the rate of diffusion of the dissolved CO2 from the liquid to the bubble. Diffusion is affected by the temperature and pressure of the gas above the liquid. 4. The speed of bubbles rising to the surface. This will depend on:
• the distance they have to travel (depth of the liquid), • the size of the bubble (a buoyancy effect), • the temperature of the liquid (volume of the bubble is proportional to absolute temperature), • the viscosity of the liquid (from sugar and other salts). The presence of acids in carbonated beverages (soda, soft drink, beer, wine) will affect the viscosity and will slow bubbles down (a good thing if you want bubbles to last longer). The strong hydrogen bonding between the citrate ion for example makes water a little more viscous and harder for the bubbles to rise to the surface. From my time as a chemist at Golden Circle cannery, I know how important it was to get the citric acid concentration just right - not only to impart the nice 'tart' taste, but to also to control bubble formation. • the invisible diffusion of dissolved CO2 through the water surface. This will be proportional to the surface area of the open container.
So, you can see, there is no single factor that determines the relationship between the rate of CO2 loss and time elapsed. It is rather complex.
Frederick Hughes, Year 12 student from Scotch College, Melbourne, sent me results for his Extended Practical Investigation for the 2024 VCE. Here are his graphs:
Method:
Soda water headspace pressure and Henry's Law The greater the pressure of carbon dioxide gas in the headspace of a bottle of soda water or soft drink, the greater the amount of dissolved carbon dioxide in the drink itself. There is a relationship between the two known as Henry's Law. In the following video I show you how to determine experimentally the headspace pressure of gas and how to calculate the concentration of dissolved gas using Henry's Law.
CAN I USE THIS FOR A STUDENT EXPERIMENT? The short answer is 'probably not'. For a good student experiment you need to show a relationship that can be plotted, such as how the pressure changes with temperature. So, you could do the experiment with bottles of soda water at different temperatures. This, in itself, is unlikely to make a great student experiment, because you can't control all of the variables. For example, you can't be certain that each bottle was prepared with the same headspace pressure. Remember that when investigating a relationship you have one independent variable (eg temperature) and one dependent variable (pressure) with all other conditions held constant. You are not able to control the headspace pressure, so it is not really a fair test. However, you could have a go and state this is a limitation of the experiment. I'd suggest you get five bottles of soda water (the small ones, about 300 mL, which are about $1 each in a supermarket). You could take each one to a different temperature, say 10oC, 15oC, 20oC, 25oC, 30oC and do the experiment on each. You could assume the five bottles were bottled at the same pressure so any difference in the headspace pressure is directly related to temperature (Gay Lussac's Law: P proportional to T, when T is in kelvin). I think if you were trying to show that the solubility of carbon dioxide gas varies with temperature you would have a problem. However, if you titrated them straight away (as in the experiment above) you would get lower acidity (and hence lower dissolved CO2) as the temperature increased.
Carbon dioxide and citric acid in soft drink. A great titration.
The problem is that soft drink has citric acid in it as well. We can measure the total acidity of the soft drink by titration, and then decarbonate it (make it 'flat') and measure just the citric acid by titration. The difference in titres is a measure of the [CO2].
This is a great experiment and will develop you analytical techniques. However, it doesn't make a good student investigation as there is no mention of relationships between variables as expected in a student experiment. You could measure the [CO2] of different drinks but that hardly forms a good reasearch question. For a good student experiment based on this technique, see the next suggestion further down. Here are some hints, some typical results, and worked calculations.
HINT 1. You can standardized the approximately 0.1 M NaOH solution against the solid acid potassium hydrogen phthalate (KHP) - a weak monobasic acid that is used as a primary standard. If you don't have KHP, you can prepare approximately 0.1 M HCL and standardize it against the sodium hydrogen carbonate NaHCO3 and then standardize the NaOH solution against the standard HCl solution.
HINT 2. When using a coloured indicator such as phenolphthalein for the titration, you need a clear uncoloured drink so you can see the end point. Lemonade works well.
HINT 3. If you are testing a coloured drink you will need a pH meter to determine the end point (pH = 8).
HINT 4. It is impossible to pipette an aliquot of fizzy soft drink as the bubbles keep expanding inside the pipette. You need to weigh out a sample on a balance. I measured out 20.00 grams and titrated that against 0.100 M NaOH using phenolphthalein indicator.
HINT 5. It is hard to get three titres exactly the same as the pouring of the sample (aliquot) into the flask disrupts the rest of the liquid. You need to think of a way to remove three samples from the container without disturbing the container too much. I measured out three samples into separate flasks at the one time. That seemed to give low spread of results (low uncertainty = high precision).
TYPICAL RESULTS. For 20.00 g aliquots of Schweppes lemonade I had titres averaging 10.60 mL. Then I flattened (decarbonated) the lemonade and measured out 20.00 g again and had average titres of 7.50 mL. This gives a citric acid concentration of 0.240% (which is about right), and a CO2 concentration of 0.00775 M which is 0.034 g/100g (0.034% w/w). This is the CO2 concentration about 40 minutes after opening. The CO2 concentration of the drink inside the can before opening would be much higher. It is likely to be about 0.37 % w/w or 0.084 mol L-1. Calculations are shown below:
CALCULATIONS FOR CITRIC ACID citric acid is a tribasic acid with three -COOH (carboxylic/alkanoic acid groups): CH2COOH-C(OH)-COOH-CH2COOH citric acid + 3 NaOH → sodium citrate + water ncitric = CNaOHVNaOH/3 = 0.100 x 0.00750/3 = 0.000250 mol citric acid in 20.00 mL mcitric = nM = 0.000250 x 192.12 = 0.0480 g citric acid in 20.00 mL % citric acid = 0.0480/20.00 x 100 = 0.240% w/v (3 sf) CALCULATIONS FOR CARBON DIOXIDE Citric acid and carbon dioxide mixture: 10.60 mL titre with 0.100 M NaOH Citric acid (degassed): 7.50 mL with 0.100 M NaOH Therefore the carbon dioxide required 3.10 mL 0.100 M NaOH Reaction: CO2 + 2 NaOH → Na2CO3 + H2O nCO2= n NaOH/2 = CNaOHVNaOH/2 nCO2=0.100 x 0.00310/2 = 0.000155 mol in 20.0 g (20.0 mL) CCO2= n/V = 0.000155 mol/0.020 L = 0.00775 M mCO2 = nM = 0.000155 x 44 = 0.00682 g in 20.00 mL %CO2 = mCO2 /20.00 x 100 = 0.034% (or 0.034 g /100 g)
HINT 3. For the theory behind decarbonation of soft drink, see the next section where I look at the less complicated situation of soda water going flat.
Rate of carbon dioxide loss in soft drink - by titration. A nice modification As I said above, measuring the CO2 concentration is only useful for a student experiment if it is a part of a bigger experiment with a valid research question. It would be interesting to see how fast soft drink goes flat. Thus, a question could be asked: what is the relationship between the CO2 concentration of soft drink in an open container, and time elapsed?
Carbon dioxide in soft drinks - effect of temperature. More titrations. A great modification to the experiments above would be to investigate the solubility of CO2 in soda water or lemonade as a function of temperature (by titration). Results obtained by this procedure are intended to indicate a trend in the solubility of the carbon dioxide as a function of temperature. This is a redirection as the independent variable has been changed from 'time elapsed' to 'temperature'.
In aqueous solution, carbon dioxide exists in many forms. First, it simply dissolves: CO2(g) → CO2(aq) Then, an equilibrium is established between the dissolved CO2 and H2CO3, carbonic acid: CO2(aq) + H2O(l) → H2CO3(aq) The reaction is reversible; that is, the products can react to form the reactants. The 'species' are said to be in equilibrium if the rate of the forward reaction equals the rate of the reverse reaction. In this case there will be no change in their concentrations. At a pH of 5.1 - which is typical of 'carbonated water' - only about 0.5% of the dissolved CO2 exists as carbonic acid H2CO3. The other 99.5% exists as CO2(aq). Carbonic acid is a weak acid which dissociates in two steps. Step 1: H2CO3 → H+ + HCO3- Ka1 = 4.2 x 10-7 ; Step 2: HCO3- → H+ + CO32- Ka2 = 4.8 x 10-11. When titrated, all the CO2(aq) is reacted so a titration is a measure of total CO2 content: H2CO3(aq) + 2NaOH(aq) → Na2CO3 + 2H2O.
POINT 1. Students often say 'if only a tiny amount of CO2 exists as carbonic acid H2CO3, won't a titration just pick up the H2CO3 and not the vast amount of the dissolved carbon dioxide (CO2(aq))?' The answer is that as the H2CO3 gets used up in a titration reaction, more is produced as the equilibrium for the reaction CO2(aq) + H2O(l) → H2CO3(aq) shifts to the right.
POINT 2. The enthalpy changes for the various reactions will allow a prediction to be made about the effect of increased temperature (that is, when energy in the form of heat is added). The ΔH (heats of reaction) value for the reactions are as follows: CO2 (g) → CO2 (aq); ΔH = -400.0 kJ mol-1 exothermic CO2 (aq) + H2O (l) → H2CO3 (aq) ; ΔH = +21.8 kJ mol-1 endothermic H2CO3 (aq) + H2O (l) → HCO3- (aq) + H3O+(aq) ;ΔH = -35.8kJ mol-1 exothermic HCO3- (aq) + H2O (l) → CO32- (aq) + H3O+(aq) ;ΔH = -14.8 kJ mol-1 exothermic Or, in summary: CO2 (g) + H2O (l) → CO32- (aq) + 2H+(aq) ; ΔH = -428.8 kJ mol-1 exothermic When the temperature is increased the added energy shifts the equilibrium towards the left (reactants). Thus, the CO2 (g) comes out of solution in the form of bubbles when it is heated.
HINT 1. Try to do the samples from different temperatures as quickly as possible. The carbon dioxide concentration decreases with time elapsed once the bottle is opened, so you need to control time as best you can. For example, it drops by about 0.5 g/L in 30 minutes after opening.
HINT 2. Normally, you'd aim for titres of approximately the same as the aliquot being used (20 mL). When using 0.1 M NaOH you may find the titres are only half that or less. You may like to use 0.05 M NaOH instead.
My thanks to Year 12 Chemistry student Codi Baker-Lahey from St Andrew's Anglican College, Sunshine Coast, Queensland, Australia for sharing the results of her EEI (2016) done under the supervision of her teacher Mrs Larsen.
Determination of total carbon dioxide by titration. Another marvellous modification. One way of measuring CO2 in soft drink involves heating the beverage in a flask and capturing the CO2 in a balloon placed over the top (that contains an excess of sodium hydroxide solution). The evolved carbon dioxide gas that passes into the balloon is absorbed and converted into an equivalent amount of sodium carbonate. The resulting mixture consisting of the excess sodium hydroxide and sodium carbonate can be titrated with standard HCl.
Titration to the first colorless phenolphthalein endpoint neutralizes the excess sodium hydroxide and converts all of the sodium carbonate into sodium bicarbonate. Methyl orange indicator is added and the titration is continued to the second endpoint (where methyl orange changes colour) as the acid converts the sodium bicarbonate to water and carbon dioxide. The difference in milliliters between the first and second endpoints is used to calculate the carbon dioxide present in the sample or the grams of substance being sought.
As a former industrial chemist at Golden Circle Cannery (soft drink manufacturer) I tried this method: I poured 100mL of chilled soft drink into a chilled 500mL conical flask and added a magnetic stirring bar. Then I poured exactly 40 mL of 1.00M NaOH into a balloon and immediately stretched the balloon over the neck of the flask - being careful not to spill any of it into the flask. If the balloon is positioned as illustrated in the photo above there is no danger of the NaOH entering into the reaction flask.
I stirred the flask at slowly first, then after the vigorous fizzing has ended, I continued with greater agitation for 15 minutes and then turned it off and allowed the flask to stand overnight. In the morning the balloon had collapsed - demonstrating the absorption of the CO2 by the NaOH.
I carefully transferred the contents of the balloon into a 250 mL conical flask and rinsed the balloon out with some distilled water. Then I titrated the solution (using 4 drops of phenolphthalein indicator) with standard HCl in the burette (approx 1M) until it reached a colorless end point. This titre (V1) was recorded. Then I added four drops of methyl orange (that turns yellow when added) and completed the titration until an orange color was obtained. I recorded the volume (V2) used to the second endpoint.
It can be shown that the difference in titres (V2-V1) is related to the amount of NaHCO3 produced which is equal to the amount of CO2 produced, that is: (V2-V1) x CHCl = n CO2 = mCO2/MrCO2.
For Schweppes Lemonade I had the following results using 1.09M HCl: V1 = 11.1 mL, V2 = 15.6 mL, V2-V1 = 4.5 mL. • (V2-V1) x CHCl = n CO2 = m(CO2)/M(CO2) • 4.5/1000 x 1.09 = m/44 • m(CO2) = 0.196g (in 100mL beverage) = 1.96 g/L (1L gas per 1L of beverage). • The correct value is probably 3L(gas)/1L(beverage), or 3:1 carbonation.
This method is based on one reported by Crossno, Kalbus & Kalbus from California State University in the Journal of Chemical Education V73(2), Feb 1996, p175. It is now available on-line here. If that link gets broken, download it off my site here.
Table sugar 'sucrose' is a disaccharide made up of a glucose and fructose molecule joined together by a glycosidic linkage. Hydrolysis breaks the glycosidic bond and converts it into glucose and fructose:
C12H22O11 + H2O + H + → C6H12O6 + C6H12O6 + H +
Hydrolysis is, however, so slow that solutions of sucrose can sit for years with negligible change. In sucrose hydrolysis studies, the main way to measure the extent of the reaction is by using a 'polarimeter' which measures the amount of rotation of polarised light. Schools just don't own polarimeters but there is a simple method that involves a simple and inexpensive blood glucometer (see photo left). The test sensors are sophisticated high-tech products with integrated nanoscaled membranes and detectors. And they only cost about $17.
There are two main ways of hydrolysing sucrose:
1. ACID HYDROLYSIS - This method involves the addition of weak acids, such as tartaric acid or citric acid (lemon juice). Likewise, gastric (stomach) acidity hydrolyses sucrose. Dependent variable: glucose concentration Independent variables: sucrose concentration, pH (using weak acids), temperature (rate of inversion increases exponentially with temperature). Method: I'd start with a 10g/100mL sucrose solution, temperature 25°C, pH7. Measure the glucose every 10 minutes with the glucose meter.
2. ENZYMES - This method involves the adding the enzyme sucrase (invertase) as found in baker's yeast. Controlled variables: Baker's yeast suspension of 0.5g/100mL Method: I'd also start with a 10g/100mL sucrose solution, temperature of say 25°C, pH7. It would also be good to add some nutrient so a Sorensen buffer (NaH2PO4 0.066 M, K2HPO4 0.066 M) made to the various pHs would be handy. Measure the glucose every 10 minutes with the meter.
NOTE: if you'd like to measure the glucose more accurately (by titration) then see the suggestion below about the ripening of bananas.
The electrical conductivity of aqueous solutions has been extensively studied and reviewed for the past several decades. However, most of the theoretical treatments and experimental investigations have been directed toward systems containing only a single cation/anion pair. There are lots of data tables around showing the conductivity of aqueous solutions as a function of their concentration. For example the conductivity of a 5% (w/w) potassium nitrate solution is 58 μS/cm but a 10% solution is just 96 μS/cm (not double as you might expect). The same is true of sodium nitrate 52 uS/cm for a 5% solution but 92 for a 10% one.
The point is that conductivity is not a linear relationship with concentration. Anyone can just look up the tables though. However, there seems nothing published on mixtures. For example, if you mixed together equal volumes of KNO3 and NaNO3 would the resultant conductivity be the average (96 + 92)/2 = 94 μS/cm? You could also argue that each solution has its volume doubled so is half the original concentration. Thus, you could just add the two 5% values and get 58 + 52 = 110 μS/cm. A good EEI would be to explore the additivity of conductivities of aqueous ionic solutions. I can't wait to try it when I get back to school. There is an very complex article by Professor Sharygin and co-workers from the University of Delaware on a related topic (J. Phys. Chem. B 2001, 105, 229-237). Download it here if you're game.
Imagine if you were making solutions of electrolytes in water for use in dialysis and a precipitate formed. What a mess you could be in. The quality of the water used in the mixing of the electrolyte and the maintenance of dialysis systems is also critically important and this can be measured by conductivity.
Chemical constituents of the water can change the ionic composition of the dialysate thus altering the concentration gradient in the dialyzer; react with constituents of dialysate or blood changing the chemical composition of the dialysate prescription or generating unwanted precipitates. This is just to highlight how important knowing how conductivity changes when precipitation occurs.
A good EEI can be made out of a fairly typical 1st year university chemistry prac. I would take 50 mL of 0.1 M copper (II) nitrate and place it in a beaker with a conductivity probe. Then I would add 0.1M sodium carbonate solution from a burette and note the conductivity as the carbonate was added. A plot of volume of carbonate added vs conductivity would be fascinating. Of course, as the carbonate is added it causes copper carbonate to precipitate so the copper ions and carbonate ions are removed from solution. The conductivity should decrease until the equivalence point (50 mL of each). Here's a graph of the theoretical calculations.
The Queensland chemistry syllabus (2025) lists the subject matter for titrations in general as:
In chemistry the term "acid-base titrations" normally means:
The syllabus confuses matters by using "acid-base titrations" to refer to the second type only (coloured indicator ones). There are other forms of "colorimetric titrations" such as any titration that uses a coloured indicator (eg potassium chromate coloured indicator for a precipitation titration).
Here's a video I made discussing student experiments on conductometric acid-base titrations:
It is usual when discussing titrations to use the abbreviations SA, SB, WA, WB as shown above. When describing a particular titration is is usual to state the 'analyte' first (the solution that goes in the flask), followed by the 'titrant' (the solution in the burette). For example SA/WB means the analyte is a strong acid (SA) is placed in the flask, and the titrant is a weak base (WB) placed in the burette. Here are the six combinations. Note that we do NOT discuss a titration between a WA and WB as it is not required by the syllabus.
Analyte (in flask)
A conductivity titration graph traces the conductivity of the analyte (in the flask) as titrant (in the burette) is added. Graphs are discussed in the YouTube video above. Let's look at theoretical graphs of each of the six possibilities. Note: these are drawn so that they are easy to understand. In practice they are not so straight.
1. STRONG ACID ANALYTE + STRONG BASE TITRANT
4. STRONG ACID ANALYTE + WEAK BASE TITRANT
Initially: The flask contains the strong acid HCl. As it is strong it is fully dissociated: HCl (aq) → H+(aq) + Cl-(aq). This means the conductivity is very high (about 35 mS/cm). Before the end point: As the titrant NH3 is a weak base it is only partly dissociated in aqueous solution: NH3 + H2O → NH4+(aq) + OH-(aq). As it is added to the acid, a neutralisation reaction occurs: H+(aq) + Cl-(aq) + NH4+(aq) + OH-(aq) →NH4+(aq) + Cl-(aq) + H2O. The H+(aq) + OH-(aq) ions are removed in equal amounts from the solution to form water. As the titration proceeds the H+(aq) ions removed from the analyte solution are replaced by an equal amount of NH4+(aq) from the titrant solution. The small and very mobile H+(aq) ions are being replaced by the larger and less mobile NH4+(aq) ion. This means the conductivity decreases. The conductivity also decreases because the volume of liquid (the solvent, water) is also increasing. At the end point: All the H+(aq) + OH-(aq) ions have reacted (in equal amounts) so no more neutralisation can occur. This is where the conductivity is at a minimum and is called the end point (20.0 mL - which indicates the equivalence point). The only ions present are NH4+(aq) + Cl -(aq) ions (as well as water H2O). In effect, we just have a 0.05M NH4Cl solution in the flask. After the end point: No more neutralisation occurs. However, as more titrant NH3 is added the conductivity increases a small amount. This is because NH3 is weakly dissociated so the ions NH4+(aq) + OH-(aq) are still being added but in small amounts.
5. WEAK ACID ANALYTE + STRONG BASE TITRANT
Initially: The flask contains the weak acid CH3COOH which is partially dissociated thus: CH3COOH → H+(aq) + CH3COO-(aq) but most molecules remain as CH3COOH. There are very few ions present so the conductivity is very low. It is actually lower than shown on the graph. In fact a 0.1M solution of acetic acid has a conductivity of about 0.5 mS cm-1.
Before the end point At the start: At the very start, as NaOH is added to the weak acid in the flask, a neutralisation reaction occurs: CH3COOH + Na+(aq) + OH-(aq) → Na+(aq) + CH3COO-(aq) + H2O. The formation of the CH3COO-(aq) ion represses further dissociation of CH3COOH by the common ion effect so hardly any H+(aq) + CH3COO-(aq) are formed. We see this as a small downturn in conductivity at the start caused by the increasing volume of solution.
As the titration proceeds But very soon the conductivity increases on adding more NaOH as the NaOH neutralises the un-dissociated CH3COOH to form CH3COONa (sodium acetate) which is a strong electrolyte and thus fully ionized. This increase in conductance continues right up to the end point. At the end point: All the H+(aq) + OH-(aq) ions have reacted (in equal amounts) so no more neutralisation can occur. This is where the conductivity is at a minimum. This is the end point (20.0 mL - which indicates the equivalence point). The only ions present are Na+(aq) + CH3COO-(aq) ions. In effect, at the end point we have a 40 mL solution of 0.05 M sodium acetate which has a conductivity of about 4 mS/cm. After the end point: As more titrant Na+(aq) + OH-(aq) ions are added, the conductivity increases (but no more neutralisation occurs). The conductivity increases as more and more highly conducting Na+(aq) and OH-(aq) are added.
Initially: The flask contains the weak base NH3 (ammonia) partly dissociated in aqueous solution: NH3 + H2O → NH4+(aq) + OH-(aq). There are very few ions present so the conductivity is very low. It is actually lower than shown on the graph, in fact it is 0.5 mS cm-1. It is exaggerated on the diagram for clarity.
Before the end point At the start: At the very start, as HCl is added to the weak base in the flask, a neutralisation reaction occurs: H+(aq) + Cl-(aq) + NH4+(aq) + OH-(aq) → NH4+(aq) + Cl-(aq) + H2O. The H+(aq) reacts with an equal amount of OH-(aq) ions to form water. The formation of the NH4+ion represses further dissociation of NH3 by the common ion effect so hardly any more NH4+(aq) ions are formed. We see this as a small downturn in conductivity at the start.
As the titration proceeds But very soon the conductance increases on adding more HCl as the HCl reacts with the un-dissociated NH3 to form NH4Cl (ammonium chloride) which is a strong electrolyte and thus fully ionized. This increase in conductance continues right up to the end point. At the end point: All the H+(aq) + OH-(aq) ions have reacted (in equal amounts) so no more neutralisation can occur. This is where the conductivity is at a minimum. This is the end point (which indicates the equivalence point). The only ions present are NH4+(aq) and Cl-(aq). At the end point (20.0 mL) all we have in the flask is a 0.05 M solution of ammonium chloride. After the end point: The conductivity increases because we're adding highly conductive H+(aq) + Cl-(aq) ions.
When investigating galvanic cells there are two approaches that can be taken: open circuit, or closed circuit.
1. OPEN CIRCUIT. This is where the cell is not being discharged (that is, not connected up to an external device such as a light bulb, resistor, motor etc). This is by far the most popular type for students to investigate as it is much easier to control the variables. In this situation, the only external device across the anode and cathode is a voltmeter or digital multimeter (DMM) used to meassure the cell potential difference (the voltage, or emf). The current through the external circuit will be extremely low as the electrical resistance of the meter is so high. Numerically, the resistance of the meter is typically 10 MΩ (megohm), or 10 million ohms, so for a cell potential difference (voltage) of 1.0 V, the current, I, is equal to V/R (by Ohm's Law). Hence I = 1 volt/10 000 000 ohm = 1 x 10-7 A (0.1 microamp, 0.1μA). This means that only a small amount of the cell's chemical species are reacting.
Practical modifications for an open cell There are a couple of suitable modifications that can be made to an open circuit cell. In terms of the Queensland (QCAA) Chemistry Syllabus these 'modifications' are classified as 'redirections' because a certain natural factor (temperature, concentration) affected results and its relationship to the original variables needs to be assessed. The modifications are:
* the effect of temperature on the cell potential difference (voltage, or emf) * the effect of concentration changes on the cell potential difference
It is easy to think of other changes but most of them have no impact on the voltage of an open circuit cell. The changes that have no impact on an open cell voltage are things like the number of salt bridges, the concentration of salt bridge ions, different salt bridge compounds (eg KCl vs KNO3 vs NH4NO3 vs NaCl), the submerged surface area of the electrodes or salt bridge. Look - here's an example:
The size of the current is negligible thus the motion of the ions is small. Even though some ions move faster than others the influence of a potential difference, this is of no consequence for an open circuit as the ions are not moving much. As I said, these may have an impact with cells under load, but not with an open circuit cell. Don't bother with them unless your experimental design is based on a null result. This can be useful but is unlikely to provide data suitable for analysis and discussion at a high level.
2. CLOSED CELL (UNDER LOAD). This is where the cell is being discharged, usually by some external 'load' device placed across (between) the anode and cathode. This will be a device that allows a moderate current to flow through the external circuit. Examples are light bulbs (torch bulbs), or a resistor (eg 100 Ω). In my experience, resistors are better than bulbs as bulbs heat up more and the resistance changes and ae said to be 'non-ohmic'. For a 100 Ω resistor across a 1.0 V cell, a current of approximately 0.001 A (1 mA) will flow. This means an appreciable chemical reaction is occuring. It is about 10 000 times the current flowing into the voltmeter.
There are a couple of suitable modifications that can be made to cell under load:
* the effect of the load resistance on the cell's voltage and current * the effect of salt bridge area, or electrode area, on voltage * cell voltage and current during discharge as a function of time
Let's look at some possibilities for open circuit cells first, and later, for cells under load.
WHAT VOLTMETER SHOULD I USE?
The most common type of voltmeter in schools today is the digital multimeter (DMM). The analog (printed scale) type will still be around but they are no longer as common as the DMMs have become so cheap.
An analog voltmeter has a printed scale. This one has connections to the 3 V scale so is reading 1.05 ±0.05V. They are fairly cheap ($25) but students tend to burn them out.
HOW ACCURATE ARE DIGITAL MULTIMETERS?
Students often get cell voltages lower than the theoretical value and so often in their 'improvements' section of the report mention getting a more accurate voltmeter. I've tested the cheapest of the DMMs and they are extremely accurate so it is not really an issue.
You are bound to have made a Daniell Cell while doing classwork on galvanic cells. It is written Zn(s)/Zn2+(aq) // Cu(s)/Cu2+(aq) which shows that it is made up a zinc half cell (anode) and a copper half cell (cathode), joined by a salt bridge. The standard electrode potential, Eo, is 1.102 V (measured at 25°C, 298 K). The spontaneous reaction is: Zn(s) + Cu2+(aq) → Zn2+(aq) + Cu(s).
THE ANION PROBLEM - WHICH ANIONS SHOULD I USE? No-one talks about which anions (negative ions) to use. That is, should you use sulfates, SO42−, or nitrates, NO3−, and does it really matter? It does matter if you want to get an experimental cell voltage close to the theoretical voltage. For the Daniell Cell containing the cations Zn2+(aq) and Cu2+(aq), the standard cell potential, Eocell is 1.10 V but certain combinations of anions will prevent you getting close to that. If you look at the cell half-reactions, at first sight, anions do not seem to take part in these reactions and most textbooks treat them as if they have no effect. But they do. The anions give rise to an additional potential difference in several ways. Two of the most common are:
The metal-electrolyte boundary where they inhibit the anodic and cathodic reactions due to the way they affect the surface layer (the 'diffusion layer') and prevent ions from arriving at the cathode, or leaving the anode; The liquid junction boundary - the boundary between two electrolytes of differing nature and/or concentration - where differing rates at which the anions move thorough the solution - known as 'the 'diffusivity' - create an imbalance in charge and hence a potential difference. We use a salt bridge with a high concentration of ions (of equal diffusivity such as K+(aq) and NO3−(aq)) to reduce this liquid junction potential.
The metal-electrolyte boundary where they inhibit the anodic and cathodic reactions due to the way they affect the surface layer (the 'diffusion layer') and prevent ions from arriving at the cathode, or leaving the anode;
The liquid junction boundary - the boundary between two electrolytes of differing nature and/or concentration - where differing rates at which the anions move thorough the solution - known as 'the 'diffusivity' - create an imbalance in charge and hence a potential difference. We use a salt bridge with a high concentration of ions (of equal diffusivity such as K+(aq) and NO3−(aq)) to reduce this liquid junction potential.
Results: Here are the experimental voltages I get when I make a Daniell Cell with different combinations of anions using a saturated KNO3 salt bridge. The ones with least error are shaded. The percentage error to the accepted value of 1.10 V is also shown.
It doesn't seem to affect the results of any of the experiments that follow, but just be aware you do get lower voltages than expected.
PREDICTING THE EFFECT OF TEMPERATURE The Daniell Cell reaction is exothermic and has a heat of reaction, ΔH, of −213.6 kJ/mol. We can think of the thermal energy as being a product of the reaction:
RESULTS Here are my results in graphical form. I used a 1.0 M zinc sulfate solution and a 0.001 M copper sulfate solution with a salt bridge of saturated potassium nitrate. Hence, [Zn2+] = 1.0 M, and [Cu2+] = 0.001 M. That is, [Zn2+] > [Cu2+] and the ratio [Zn2+] / [Cu2+] is 1.0/0.001 = 1000. This ratio is called the Q-ratio ("Q" for quotient, short for reaction quotient). I've converted the temperatures to kelvin (K) to see a more useful equation for the linear trendline. In a student experiment you would do replicates at each of the six data points and indicate the spread (uncertainty) at each of the six using error bars.
NOTE 1. The relationship between voltage and temperature is given by the Nernst Equation
- where R is the universal gas constant and is equal to 8.31 J K-1 mol-1, T is temperature in kelvin (K), n is the number of electrons transferred for every ion (n = 2), F is Faraday's constant = 96485 C mol-1, and Q is the activity constant or reaction quotient = [Zn2+]/[Cu2+] for a Daniell Cell. The term ln Q stands for the natural log of Q, or logeQ.
Sample calculation Question. Calculate the potential difference of a Daniell Cell made of [Zn2+] = 1.00 M, and [Cu2+] = 0.001 M at a temperature of 20oC (293 K). Solution
If you want to use base 10 logarithms the equation becomes:
Sample (using data from question above):
For equimolar solutions of zinc and copper, that is [Zn2+] = [Cu2+], the value of Q = [Zn2+]/[Cu2+] is 1 and because the value of ln(1) is zero, that whole term in the equation (RT/nF × ln Q) reduces to zero so there will be no effect of a temperature change. This is also true for log10 calculations. If [Cu2+] > [Zn2+] then you will get an increase in voltage with an increase in temperature.
SUMMARY
Q Ratio: [Zn2+]/[Cu2+]
We can use Le Chatelier's principle to predict the effect of increasing temperature. For a cell where [Zn2+] > [Cu2+], as the temperature is increased the equilibrium shifts towards the left (to use up the added heat) and so the voltage will decrease. However, the behaviour of the system is far more complex than can be predicted by Le Chatelier's principle alone. This is because the Eocell value is also affected by temperature. So, Le Chatelier's principal only takes into account one aspect of the equilibrium and not the other. Let's just stick with the Nernst equation to predict changes to the voltage with chnaging temperature.
THE NERNST EQUATION - DO I NEED TO USE IT? You, like many students, may want to compare your experimental results with the accepted values. So as to be able to calculate absolute and percentage error you will need to generate secondary (theoretical) data in order to compare with your own primary data. However, this can be done without putting values into the Nernst equation. You do not need to mention the Nernst Equation in your experimental report, even to get top marks. The Nernst Equation is not listed as subject matter in the Queensland Chemistry syllabus so it would not appear in the External Exam, and you would not necessarily have to use it in your experimental discussion. You are still able to get full marks without referring to it. Here are some hints if you are not going to use it:
* There are a number of simulators online such as this one: https://web.mst.edu/~gbert/Electro/Electrochem.html. Also the company Newbyte has one for this purpose. Using simulators completely avoids any need to even mention the Nernst equation, and discussion about not being at equilibrium and the direction the reaction will tend to go in. * Additionally, there are online calculators that use the Nernst equation and calculate it for you: http://calistry.org/calculate/nernstEquation. If you use this source of secondary data you sould mention the source and how it is obtained as a reference, but there would be no need to go into anything in depth about it. * Have a look at the 'high' sample student experiment provided on the QCAA website to see how the analysis is done if you want to refer to the Nernst Equation. Remember - it is only a sample and is provided to help you with your decision making.
* Additionally, there are online calculators that use the Nernst equation and calculate it for you: http://calistry.org/calculate/nernstEquation. If you use this source of secondary data you sould mention the source and how it is obtained as a reference, but there would be no need to go into anything in depth about it.
* Have a look at the 'high' sample student experiment provided on the QCAA website to see how the analysis is done if you want to refer to the Nernst Equation. Remember - it is only a sample and is provided to help you with your decision making.
HINTS FOR THE EXPERIMENTAL PROCEDURE HINT 1. You will need to make the concentrations different (eg 1.0 M and 0.01 M). Depending on which one you make bigger, you will get an increase in voltage with increased temperature (when [Zn2+] < [Cu2+]), or a decrease in voltage with increased temperature (when [Zn2+]>[Cu2+]). Just check the Nernst equation. In the graph above, I used [Zn2+] =1.0 M and [Cu2+] = 0.001 M. Thus, the voltage will decrease as the temperature increases. I prefer to keep [Zn2+] large and [Cu2+] small because the voltage will drop with a temperature increase so you're not trying to push the voltage higher against other losses in the cell. However, I've seen great results from students who go the other way and use 3.0 M [Cu2+] and 1.0 M [Zn2+].
HINT 2. The bigger you make the difference in the concentrations of Zn2+ and Cu2+, the bigger will be the second term of the Nernst Equation (RT/nF x lnQ) and so the bigger will be the change in voltage with temperature change. Here's an example to show what I mean. If the Q ratio ([Zn2+] /[Cu2+] is small, eg 2: ([Zn2+ = 2.0M] and [Cu2+ = 1.0M], lnQ is 0.693 and a change in temperature has almost no measurable effect (at 278K (6oC, E = 1.0916 V, and at 333K (60oC), E =1.0900 V. That's a change of 0.0016 V. That’s for the biggest temperature range safely possible in the lab. A digital multimeter (DMM) will struggle to show a difference especially when the scale reading uncertainty for a DMM would be +/- 0.001, which would be a 60% scale reading uncertainty. But, if you make the concentration difference greater, for example, [Zn2+ = 1.0 M] and [Cu2+ = 0.001 M], Q is very large (1.0/0.001 = 1000) so lnQ is 6.9 and thus a change in temperature from 6oC to 60oC gives a voltage change of 0.016 V which is 10 times greater than previously. A DMM can display that change with less scale reading uncertainty (6%).
HINT 3. Be careful with solubilities of zinc compounds. Anhydrous zinc sulfate, ZnSO4, has a solubility of about 58 g/100 mL at 20oC or about 3.5 mol/L. However, zinc sulfate heptahydrate ZnSO4.7H2O has a solubility of only 54 g/100 mL which is about 1.87 M. So be certain about which type you have. The label on the main container will state the molar mass. If it says MW 161.47 then its the anhydrous form. If is says MW 287.54 then its the heptahydrate form. Zinc nitrate on the other hand is much more soluble. It is usually supplied to schools as the hexahydrate, Zn(NO3)2.6H20, which has a molar mass of 297.5 g/mol, and has a solubility of at 184.3 g/100 mL (6 mol/L). When developing your method check the label for the exact formula and molar mass. The anhydrous form of zinc nitrate, Zn(NO3)2, is not readily available and it has a molar mass of 189.4 g/mol.
HINT 4. I suggest you keep the Cu2+ concentration as low as possible, but not too low as the conductivity depends on the concentration of ions. I've found that a [Cu2+] of 0.001 M or 0.0001 M works well. When [Zn2+] = 1.0 M, and [Cu2+] = 0.0001 M, the activity constant Q = [Zn2+]/[Cu2+] = 1.0/0.0001 = 10,000. This gives an lnQ = 9.2 so the impact of temperature on voltage should be evident when using school voltmeters or multimeters. You can use copper sulfate or copper nitrate as the electrolyte.
HINT 5. If you're making the concentration of the Cu2+ high, be aware of the solubilities. Copper sulfate is usually supplied to schools as the pentahydrate, CuSO4.5H2O, which has a molar mass of 249.7 g/mol and a solubility of 32 g/100 mL (1.28 M). Copper nitrate on the other hand is far more soluble. It usually is supplied to schools as the trinitrate, Cu(NO3)2.3H2O. It has an Mr = 241.6 g/mol and an incredible solubility of 267 g/100 mL. That's about 11 M. It isn't very expensive (about $69 for 500 g) but would be very viscous at high concentrations and would affect your results. I wouldn't go over 3 M.
HINT 6. While it seems logical to make the [Zn2+] as large as possible there are problems. The maximum concentration for zinc sulfate is about 1.8 M, but at this concentration the solution is quite viscous (thick). This tends to impede the motion of ions in solution which gives unrelaible results. It is said to lower the 'activity' of the zinc ions. My suggestion is to use 1.0 M zinc sulfate as a maximum.
HINT 7. Keep the volumes of electrolytes in each half-cell as small as possible. Schools have to watch their budgets. You should not need to go past about 100 mL for each half cell. I use small (60 mL) specimen jars (about 35c each in bulk, or $1 singly from Chemist Warehouse) with just 50 mL of solution. They are better than beakers as they have straight sides and it is easy to clip the electrodes to the side. Some schools do it on a microscale with less than 1 mL per half cell.
HINT 8. Get rid of the copper and zinc oxide coatings by adding acid. If you find you're results are going in the opposite way to what is expected it is most likely that a coating of zinc or copper oxide has formed on the surface of the electrodes preventing a reliable reaction. When making up the copper ion solution from copper nitrate or copper sulfate, the pH of the final solution will be about 3.2. At pH >4 the copper ions react with OH- ions and dissolved oxygen in the water to form the insoluble (precipitate) copper hydroxide Cu(OH)2. This coating on the surface of the copper electrode interferes with the functioning of the cell and you may end up with voltages lower than you expect. If you have troubles with your prac, check the pH of your electrolyte solutions. Add a few drops of 0.1 M sulfuric acid H2SO4. For the zinc sulfate and zinc nitrate solutions - they will have a pH of about 5.6, so you can add say 9 drops of 0.1 M H2SO4 to 100 mL of solution to get the pH just under 4.0 if you want to.
HINT 9. Contamination is a big problem with this prac. You need to use separate paper towels and sandpaper for each metal as the surface can easily get contaminated, particularly the zinc metal with copper solution. Even copper solution on your fingers when you touch the zinc metal will spoil the results.
HINT 10. How many trials do you need to do? That is, how many variations of the independent variable should you do? It is commonly stated that a 5 × 3 approach is recommended. That is, 5 variations of the independent variable (IV) with 3 repetitions of each. This is appropriate when the expected relationship is linear (as it is in this case). For an experiment where the relationship is expected to be non-linear then a 7 × 3 (or more) may be appropriate. Three replicates is usually satisfatctory to measure the uncertainty of the results.
HINT 11. If you construct a Daniell Cell and heat it up the voltage and temperature can be plotted to show their relationship (with the independent variable T on the x-axis). You could plot T values in oC if you like as I did above but it makes more sense to plot absolute (kelvin, K) temperatures. It should be linear with a negative slope (gradient) if you have constructed the cell with [Zn2+]>[Cu2+]. You can then draw a line of best fit and state the equation for the line.
HINT 12. You could have Excel calculate the coefficient of determination R2 (R squared) value. This is a measure of the 'goodness of fit' of the trendline to the data points. It is the square of the Pearson correlation coefficient, r. That being said, R2 is a measure of the precision or spread of results around the trendline. A value of R2 > 0.98 would be considered high precision (excellent). It means that the formula for he trendline has captured the data well. Another way of saying it is that the equation and trendline accurately represent 98% of the data points.
HINT 13. What thickness copper strips should I use? Copper sheet comes in a variety of thicknesses from 0.1 mm (foil) through to 1 mm sheets. All sizes work but if you want to have copper strips that you can use over and over, and use for electrolysis experiments, then a thickness of 0.5 mm is probably wiser, but anything up to about 6 mm is okay.
HINT 14. STOP HERE! Most students don't take the analysis any further once they show that the relationship is linear and discuss the uncertainty of the results. That is the extent of the analysis in the QCAA 'high' sample. However, you may like to take it further (but you don't have to to get full marks). Here we go.
The equation for the line given by Excel in my graph above was: y = -0.0007 + 1.1876. The Nernst Equation could be written in the form:
- where k is the constant (R/nF x lnQ). This can be rearranged as :
Ecell = –kT + Eocell y = mx + c.
Thus, you can relate your graph's equation to the Nernst Equation and so the magnitude of the gradient, m, (0.0007) will be a measure of R/nF x lnQ, and the intercept on the y-axis, c, will be a measure of Eocell. You can also work out the theoretical value of R/nF x lnQ. For example, for [Zn2+] = 0.3 M, [Cu2+] = 0.01 M, the constant will be:
Here's the theoretical graph (below) based on the Nernst equation using [Zn2+] = 1.0 M, [Cu2+] = 0.001 M going from a temperature of 17oC to 65oC (or 290 K to 334 K). I said before that the intercept on the y-axis, c, will be the prediction for Eocell. This makes sense because when the temperature is zero kelvin (0 K), the second term of the Nernst Equation becomes zero, so Ecell = Eo − 0. That is: Ecell = Eocell. This would be irrespective of the concentrations of the electrolytes in each half cell.
When I plotted all my experimental values I got a linear graph with a gradient of -0.0003 V/K as shown by the equation. The theoretical value for Eocell for a Daniell Cell is 1.10 V as we know from earlier.
I've put the theoretical and experimental results together on the graph below so you can see how they compare. Students don't generally work out the theoretical relationship and add that to the graph. That is just icing on the cake. The criteria for analysing the experimental relationship is in terms of the experimental results only.
The absolute error, Ea, in the gradient is |−0.0003 − −0.0007| = 0.0004. This is a percentage error, E% of 0.0004/0.0003 × 100 = 130%. This is very high and implies a systematic error seeing the R2 value of 0.85 says that the data points do not diverge too much from the trendline. Note that as T increases, the lines diverge indicating that the error is increasing. This is no doubt a systematic error probably caused by faulty assumptions in the Nernst Equation, rather than from experimental error. I'll look at that later. The absolute error, Ea, in the intercept is |1.10 − 1.19| = 0.09, which is a measure of Eo. We can say this because as T approaches zero, the second term of the Nernst Equation approaches zero, and hence Ecell = Eocell. This is a percentage error, E%, of 0.09/1.10 × 100 = 8%, which is not too bad for this sort of chemistry experiment in a high school lab.
MORE RESULTS Here are some results from a student report where they used [Zn2+] = 3.5 M using zinc nitrate, and [Cu2+] = 0.001 M using copper sulfate. A 1.0 M KNO3 salt bridge was used. Experimental values are shown by the orange line in the graph below along with error bars to show uncertainty in the results.
Here is the same graph as above but with theoretical values from the Nernst Equation added. These are shown by the blue line in the graph.
What galvanic half-cells should I avoid?
There are a number of other half-cells that give good results. For example Mg(s)/Mg2+(aq) and some others listed below. However, there are two that should be avoided in high school chemistry. The first is Pb(s)/Pb2+(aq) as it is banned in most schools as it is too hazardous. The other is the aluminium half-cell Al(s)/Al3+(aq) - because it doesn't give values as expected. The Eo for Al(s)/Al3+(aq) half cell is about -1.68 V. However, consider an aluminium half-cell in conjunction with the copper half-cell Cu(s)/Cu2+(aq). If the overall reaction was 2 Al + 3 Cu2+→ 3 Cu + 2 Al3+ as expected, we should get a cell potential of +1.34 V. In practice, using 1 M solutions of both, we get about +0.83 V. The reason is that this is not the reaction that occurs because the aluminium electrode is in contact with air and is always covered by a thin, continuous and colorless layer of aluminium oxide Al2O3 (or alumina) that prevents any aluminium atom from reacting with the aqueous solution it is dipped in. If you scratch this outer surface, the O2 molecules from air will immediately react and regenerate this protective alumina layer.
So in the Al/Cu cell, the metallic Al atom does not react. The overall reaction is due to water in which the first a few molecules of water cross the alumina layer through pores or by diffusion and react with aluminium metal according to: 2 Al + 6 H2O → 2 Al(OH)3 + 3 H2
This reaction happens as soon as the aluminium plate is dipped into water, and stops quickly. So a rather small amount of H2 is adsorbed on the metallic surface under the oxide layer. It does not produce any bubbles. Later on, when the plate is coupled with another electrode, this adsorbed hydrogen produces the anodic half-equation: H2→ 2 H+ + 2e-
We can work out the cell voltage for this using the Nernst equation:
But its potential is difficult to calculate because both the pressure p(H2) is unknown and must be rather small; and the concentration [H+] must also be small (with an unknown pH < 5). On the other electrode, copper, there's no problem: the half-reaction is as expected: Cu2+ + 2 e-→ Cu.
So the cell is not Al/Cu but H2/Cu and the overall cell potential is not easy to calculate. It is best to replace the aluminium by another metal like Zn or Mg.
I'm sometimes asked if a good modification would be to change the temperature of just one half-cell in a Daniell Cell. That is, leave the copper half-cell at room temperature (say 25°C) and vary the temperature of the zinc half-cell from 25°C to 80°C. The short answer is "no"; you'll just get a smaller range of voltage change, and in fact will probably be unmeasurable on your school voltmeters. Here's the reason.
We can take the Nernst equation and expand it so that the zinc and copper are seperate:
We want the biggest difference we can make for the zinc and copper concentrations so that the effect of temperature will be measurable. Thus make [Zn2+] = 3.0 M, and [Cu2+] = 0.001 M. Make the temperature range of zinc half-cell from 293K to 360K, but keep Cu half-cell at 298 K:
If we want a bigger change in Ecell then we can change temperature of both half-cells:
We can see that this is a tenfold increase in the voltage change per kelvin.
Here are the graphs: Firstly, if only the Zn has a temperature change:
The problem is the voltmeters may not be able to show any difference in the readings as the uncertainty for a voltmeter reading to 3 decimal places is +/- 0.001 V.
Secondly, if both have a temperature change:
Voltmeters will pick up this voltage change.
Lastly, we can combine them on the one graph:
You can see how much greater the voltage change is when both half-cells have their temperatures changed.
Hope this helps.
Galvanic cell - effect of temperature on a zinc/ferricyanide cell Another good cell to make and test the effect of temperature is this one:
Zn(s) | Zn2+(aq, 0.001M) || Fe(CN)63-(aq, 0.005M), Fe(CN)64-(aq, 0.005M) | Pt
Set it up and measure the EMF over the range from say 0°C to 60°C. Be warned: potassium ferricyanide Fe(CN)63- and potassium ferrocyanide Fe(CN)64- are poisonous; use gloves when weighing them out. Make sure you do a good risk assessment and check with your teacher before starting. You may be refused permission.
The relationship between voltage and concentration is given by the Nernst Equation Ecell = Eocell − RT/nF × ln Q. The value of Q (activity constant) is equal to [products]/[reactants] (= [Zn2+]/[Cu2+]). To get a reasonable voltage change you will need to make the concentrations of the [Zn2+] and [Cu2+] have as large a difference as possible. For example, keep [Zn2+] = 3.0 M and and vary [Cu2+] from 0.001M to 0.5 M in 7 steps. As the concentration of the copper ion is increased the lnQ gets smaller so the Ecell gets greater. This makes a neat experiment. Conversely, if you choose to make [Zn2+] your independent variable and keep [Cu2+] constant, then as [Zn2+] is increased, lnQ gets larger so the Ecell gets smaller.
As it will be a logarithmic relationship you could go up in logarithmic steps: 0.001 M, 0.003 M, 0.01 M, 0.03 M, 0.10 M, 0.30 M, 1.0 M. Watch contamination when you do your serial dilutions.
The solubility of zinc sulfate heptahydrate ZnSO4.7H2O is 54 g/100 mL at room temperature and that would make a 1.8 M solution. If you use zinc nitrate trihydrate Zn(NO3)2.3H2O you can go up to higher concentrations (eg 3 M).
MY RESULTS. EXAMPLE 1 - VARYING [Cu2+] AND KEEPING [Zn2+] CONSTANT AT 1.0M
I used [Zn2+] = 1.0 M, and [Cu2+] of 0.001 M, 0.003 M, 0.01 M, 0.03 M, 0.10 M and kept the cells at a temperature of 25oC (298 K). This gave me 5 variations of the independent variable. You, as a student, would be wise to do more variations as you know the relationship will not be linear. I would suggest doing three more perhaps in the range 0.1 M to 1.0 M. I would also suggest you do a minimum of three replicates (trials) of each variation to provide evidence for your uncertainty evaluations and help reduce random error..
The equation for the graph as provided by Excel says 0.0066 ln(x) which means it is logarithmic. The 'ln' term means that it is the natural logarithm with a base of 'e' rather than base 10 that students are more familiar with. We can linearise the graph by plotting cell voltage vs ln[Cu2+].
You could also linearise the graph by plotting the log to base-10 of the [Cu2+] as shown below:
The y-intercept of 1.03 V is the predicted standard cell potential, that is, when both the concentration of Zn2+ and Cu2+ are both equal to 1.0M and hence a Q-ratio of 1.0. This is because the log(10) of 1.0 is zero so the second term in the Nernst Equation becomes zero and we are left with Ecell = E0cell.
MY RESULTS. EXAMPLE 2 - VARYING [Zn2+] AND KEEPING [Cu2+] CONSTANT AT 1.0M
Should you choose to vary the zinc ion concentration and keep the copper ion concentration constant you should get results like this. Note that the [Cu2+] is kept constant at 1.0 M. As the zinc ion concentration increases, the voltage decreases.
And the log graph will look like this:
HELP. MY GRAPH GOES THE WRONG WAY!
I often get a situation where the students say: "My graph goes the wrong way. It's the opposite of what the Nernst equation says". They wonder what went wrong.
THEORETICAL RELATIONSHIP BETWEEN VOLTAGE AND CONCENTRATION
As already mentioned, the Nernst Equation gives the theoretical relationship between voltage and concentration.
We can determine the theoretical cell potential difference (Eo) by substituting the concentrations for [Zn2+] and [Cu2+] and using 298 K for T. The values for the other constants were given earlier. Note that in the graphs below I have used the absolute value of the log and made it positive.
What you will notice is that both are linear but with different gradients. It seems that as concentration of Cu2+ increases the voltages decrease as expected but the lines appear to be converging (coming together). That suggests a systematic error in either the experimental method or in the interpretation of the Nernst Equation. As well, the y-intercept is 1.10 V for the theoretical calculation. This value is to be expected as it is the Eo value for the Daniell Cell. When the concentrations of Zn2+ and Cu2+ are equal (when [Cu2+] = 1.0 M), the ln value is zero (ln 1 = 0), so the second term of the Nernst Equation becomes zero, and Ecell = Eo.
The absolute error in Eo = |1.10 − 1.0354| = 0.06 V which is 6%. The absolute error in the gradient is |0.0125 - 0.0066| = 0.006 V-1mol-1L-1. This is a percentage error of 0.006/0.0125 x 100 = 50%. This is very high and we can speculate on the source of the error. It is to do with the assumptions made in the Nernst Equation.
ASSUMPTIONS MADE IN THE NERNST EQUATION
The main assumption made in the general interpretation of the equation is to say that the value for the reaction quotient Q can be approximated by the concentration of the ions in mol L-1. However, this is only true for very dilute solutions. When a solution is not dilute the ions are not as free to move around and behave as a less concentrated solution. The solutions we are using range from 0.01 M to 1.0 M so they are not dilute and the approximation using mol L-1 adds error to the calculation. We should replace the concentrations with 'activities'.
The activity depends on the size of the hydrated ion and the temperature. The hydrated ions of copper and zinc are similar sizes so that makes things easier. To work out the activity of a particular concentration we just multiply the concentration in mol/L by the activity coefficient gamma γ. Hence a = c x γ. Here are the activity coefficients for the various concentrations I used (source: R. A. Robinson, 'Activity'. JACS, vol. 58 (6), 1936, pp. 959-961):
The activity coefficient for Zn2+of concentration 1.0 M (as we used) is 0.041. When we use activities instead of concentrations in our calculations we get a slightly different graph - as shown below. Note that I have used the absolute value of the log:
The top line (green) is based on activities and what you'll notice is that the gradient is almost the same as the experimental values (bottom blue line). This seems to explain why the experimental gradient was different to the theoretical value using concentrations (red middle line). The error in the gradient is now just 16%. That's a lot better. However, the intercept is the same as before.
The graphs below are only theoretical. I'd say you would have fairly big error bars on the graph (if you use error bars) and once you calculate maximum and minimum gradients (if you do that) you would find that the theoretical gradient (−0.0079) will be within the range of the maximum and minimum experimental gradients. That would give you confidence to say the gradient is accurate. I've made a You Tube video on adding custom error bars to a graph so long as you have at least two data points (voltages) for each concentration. See Adding custom error bars to a graph https://youtu.be/mVS7YWENShs
In the section above, the relationship between concentration and EMF (voltage) has been discussed when the concentration of just one of the half-cell electrolytes is changed. But if you extend that to include changes in concentration for the other half cell you can get a far more interesting set of data to analyse. Admittedly you can't plot 'concentration' on the x-axis as the independent variable as it would be referring to two different chemicals (eg ZnSO4 and CuSO4). However, if you plot Q (or preferably lnQ or log10Q) the graphical relationship becomes apparent.
Here's one I did using a Daniell Cell. In one set of trials I held [Zn2+] constant at 1.0 M, and varied the [Cu2+] from 0.0001 M to 1.0 M. This gives a positive value for lnQ. In the second set of trials I did the reverse: I held [Cu2+] constant at 1 M, and varied the [Zn2+] from 0.01 M to 1.0 M. This gave a negative value for lnQ. The centre line of the x-axis is where lnQ = 0, that is, where [Zn2+] = [Cu2+] = 1.0 M. Here are my results:
When lnQ = 0 (on the x-axis) the line should cut the y-axis (voltage) at the Eo value for the cell. In this case I got 1.0736 V whereas the accepted value is 1.10 V. This gives an absolute error, Ea of |1.10 - 1.0736| = 0.026 V (or 0.03 V to 2 d.p.). This is a percentage error, E%, of 0.026/1.10 x 100 = 2.4%. The absolute error in the gradient is 0.0038 V, which give a percentage error of 30%. This is high and deserves some comment as to the likely source (a systematic error I would say).
EXPT 3. Concentration cells This is a great prac as it is simple and quick to do but allows for lots of analysis and discussion. A concentration cell is an electrochemical cell made up of two half-cells with the same electrodes and electrolytes, but differing in electrolyte concentrations. As the cell as a whole strives to reach equilibrium, the more concentrated half cell becomes diluted and the low concentration half cell becomes more concentrated via the internal transfer of ions, and the external transfer of electrons. Therefore, as the cell moves towards chemical equilibrium, a potential difference (voltage) is created.
An example for this type of cell is where the cathode consists of Cu2+ (1.0 M)/Cu and the anode consists of Cu/Cu2+ (0.10 M). In this cell, the flow of electrons from the anode to the cathode is due to the oxidation of Cu metal at the anode and the reduction of Cu2+ ions at the cathode into metallic copper.
The relationship between the concentration of the [Cu2+] in the two half cells and the potential difference is given by the Nernst Equation. The anode reaction (dilute) is the numerator (top) in the Q term, and the cathode reaction (concentrated) is the denominator (bottom). It appears as:
In my experiment I kept the [Cu2+] = 1.0 M in the concentrated cell and varied the dilute cell [Cu2+] from 0.01 M to 0.80 M.
RESULTS
2. CLOSED CIRCUIT CELLS (CELLS UNDER LOAD)
A closed circuit cell is one where there is a load across the electrodes of the cell. Instead of registering an open circuit emf (voltage) we will see the voltage under load and it will be lower. By 'load' I mean anything that draws a substantial curent from the cell.
You may have noticed this if you watch the headlights of a car when the engine is started. They dim noticeably. The starter motor draws a large current from the battery and so acts as a large load on the cells. The 12 V emf of the car's battery drops to about 10 V under load and so the headlights (in parallel) have less voltage available and so get dimmer.
With a Daniell Cell (or similar) when a load is placed across the terminals there will be a larger current coming from the cell compared to just having a voltmeter there. Thus, factors that affect resistance inside the cell will come in to play. These factors include electrolyte concentration, electrode area, salt bridge ions, salt bridge area, and polarisation. These 'internal resistance' factors will cause the displayed voltage to be lower than the open circuit voltage (with no load).
These changes make great experiments but if you are looking for modifications suitable for a Queensland QCAA 'student experiment' the changes may be getting too complicated. You should discuss this with your teacher.
HOW MUCH LOAD RESISTANCE IS NEEDED TO CAUSE AN EFFECT? A digital multimeter (DMM) is used to measure cell emf and will take some electrons from the cell to determine their energy so it can give a voltage reading. It doesn't need many electrons to do this. For example, the resistance, R, of a DMM is about 10 MΩ (10 megohm, or 10 million ohm). For a 1.10 V Daniell Cell, the current drawn by a 10 MΩ load resistance is given by Ohm's Law V = IR. The current, I, equals V/R = 1.10/(1 × 10-6) = 1.1 × 10-7 ampere (A). This is extremely small and would cause no measurable change of concentration over the course of an experiment. Even if you left the DMM connected across the cell for an hour, the concentration would change by 2 x 10-9 M. This would be unnoticeable in even a 0.0001 M solution. Hence, the DMM is not acting as a load.
A more realistic load resistance would be something similar to the resistance of the cell itself - which is in the order of a several 100 to several 1000 ohm. Let's say 1000 Ω. This would produce a current (I = V/R) of 1.10/1000 = 0.0011 A. In an hour, the concentration change of 0.000001 M in a 50 mL cell - which is not likely to have an systematic effect on the results. We could look at how the voltage of a cell decreases under load of say a 56 Ω load resistor. This will give us a measure of the internal resistance of a cell, and can enable us to work out the Eo value. I've discussed this below.
Note: Under the 'International System of Units' (SI), the physical quantity current has the quantity symbol I. It is measured in units of ampere, which has the unit symbol A. Most people abbreviate the unit to 'amps'. It is not officially correct but is commonly accepted. If you want to be accurate use 'ampere'.
EXPT 3. Electrochemical cells - effect of changing the load resistance As mentioned above, as the cell's current increases, the salt bridge will have increasing trouble providing a route for the internal migration of ions. But there are a number of places in a cell that will provide internal resistance to the flow of charge. These include the salt bridge, the electrode surfaces, and the solutions. Changing these can change the internal resistance and decrease the voltage provided by the cell.
A galvanic cell can be drawn as a source of emf in series with a resistor. The resistor represents the internal resistance of the cell.
To test this cell we need to allow it to discharge through an external resistor and measure the current and voltage as it does this. This diagram shows the setup. The variable resistor imposes a load on the cell and as the resistance is decreased more current flows from the cell and more energy is used up in the internal resistance. This makes the voltage go lower.
To perform the experiment you need to make a galvanic cell such as a Daniell Cell. If you use the same concentrations for both half cells you will not have to worry about temperature effects. You would then connect a voltmeter (DMM) across it. That will give the open circuit potential difference of the cell. Theoretically, for a Daniell Cell this will be 1.10 V but from experience it will be lower. Then you should connect a potentiometer (as a variable resistor) and ammeter in series and then connect them together across the cell. This will allow the cell to discharge through the resistor and give a current reading on the ammeter. With the variable resistance set to a maximum there should be almost no current flowing so the voltage will be at a maximum. As you turn the variable resistor to lower resistance the current will increase and the voltage will fall.
I used a 5 kΩ (5000 ohm) variable resistor and did three replicates for each variation of the independent variable (current). I went up in 50 μA steps (from 150 μA to 400 μA). The current is very small - and you'll need a DMM that measures in microampere (μA) - that is, millionths of an ampere. You need to plot current on the x-axis and voltage on the y-axis. I used 0.10 M Zn2+ and 0.10 M Cu2+ solutions (that is, the same concentration) at a temperature of 19.0oC. The open circuit voltage (no load) was 0.973 V.
Relationships
The equation linking the voltage across the resistor (V), the emf of the cell (E), the current (I) and the internal resistance (r) is given by:
voltage across external load resistor = emf voltage of cell − voltage lost across internal resistor V = E − I r V = −r I + E (rearranged) y = mx + c (linear relationship)
In this form, you should be able to see that when x is the current, I, the absolute value of gradient m equals r, the internal resistance, and the y-intercept, c, equals the emf, E, of the cell.
Results
The gradient, m, of 0.0026 is a measure of the internal resistance, r, of the cell. The current is in microampere (μA so you have to multiply the 0.0026 by 106 to get the resistance in ohm (Ω). The calculation for gradient is: Δy/Δx = 1.04/(400 × 10-6) = 2600 Ω. Just to say it again: the gradient appears as 0.0026 as it is based on the x-axis value being in microampere (10-6 A). This is always a trap and often appears in external exam questions.
Error analysis.
Gradient. I've added maximum and minimum lines of best fit to the graph. These must fit within the error bars to be legitimate. The gradients for these lines are shown in the textbox on the graph. They indicate the uncertainty associated with the gradient. Remember, when doing the calculations the curent is is microampere (10-6 A). The uncertainty, δ, is: (xmax − xmin)/2 = (2800 − 2290)/2 = 255. We can say the gradient has a value of 2570 ± 255 Ω. This can be expressed with the percentage uncertainty, δ%, as 2570 Ω ± 9.9%.
Eo accuracy. The y-intercept, c, is the experimental (or observed) value, xO, for Eo for the cell. In this case the experimental value is 1.0485 V (1.05 V to 3 s.f.). The accepted value, xA, for a Daniell Cell is 1.10 V. Thus there is an absolute error, Ea, of |1.10 − 1.048| = 0.052 V. The percentage error, E%, is Ea/xA × 100 = 0.048/1.10 × 100 = 4.7%. These are a measure of the accuracy of the experiment.
Eo uncertainty. The range of values for the y-intercept using the maximum and minimum trendlines give a measure of the uncertainty associated with the y-intercept (or Eo). In this case the range is from 0.94 V to 1.12 V. The uncertainty, δ is: (max − min)/2 = (1.12 − 0.94)/2 = 0.090 V. This can be expressed as a percentage uncertainty, δ%, = 0.090/1.10 × 100 = 8%.
Interpretation of Eo value. The range for Eo of 0.94 V to 1.12 V includes the accepted value of 1.10 V. We can thus say that within the limits of experimental error, the method gives an accurate determination of Eo for the cell. The complete formulas for the relationship can be written as:
E = (−0.0026 ± 0.0003)I + 1.0485 ± 0.090 V
E = −0.0026 I ± 9.9% + 1.0485 ± 8% V
Metals are able to conduct electricity because they have mobile charge carriers - namely electrons. However, in electrolytes it is not electrons that are the charge carriers but the ions in solution. These ions - mainly Zn2+, Cu2+, SO42- - are needed for conduction and it seems logical that the greater the concentration of a particular ion, the greater the conductivity. This is borne out by tables of conductivity of various ions at different concentrations. But with galvanic cells the situation is more complex. Conductivity in cells like the Daniell Cell, relies on the oxidation of metal atoms to ions, and the reduction of metal ions to the metal atom. Layers of solvated ions form on the surface of the metal and these polar molecules can inhibit conductivity. In summary, the internal resistance provided by the metal atoms, cations and anions, and solvent molecules may only be partly determined by conductivity of the electrolyte. There may be other factors.
We can measure the internal resistance of cells with various concentrations of electrolytes using the method used above. I tried this using the same Zn2+ and Cu2+concentrations in each half cell, from 0.01 M to 1.0 M.
You can see that internal resistance (gradient) decreases as concentration increases. This is to be expected. The graph below shows the relationship. It appears to be fairly linear (R2 = 0.9908) but an exponential one is a better fit.
EXPT 5. Internal resistance of a lemon battery A common form of zinc/copper cell can be made using the citric acid solution in a lemon as the electrolyte. At the zinc anode, Zn is oxidized to Zn2+, while at the cathode, the H+ ions in the citric acid are reduced to molecular hydrogen: 2H+ + 2e- → H2(g)
You should get a open cell voltage of about 0.76 V under standard conditions (1 M solutions of reactants and products, 25oC). However, you may get more depending on the vagaries of your lemon and the setup. I regularly get about 0.9 V and the Nernst Equation helps me explain that. The current is very small (0.1 mA - 1 mA) so don't expect it to be a useful power source. Of course you could use a magnesium anode and get 1.6 V. It still won't be enough to light up a torch bulb but you can get a LED to glow.
Here is the setup I used, and a graph of the results. The separation distance and surface area of the electrodes will affect the internal resistance.
EXPT 6. Internal resistance of new and old batteries You can also measure the internal resistance of commercial batteries using the same method. I used alkaline batteries which have the following half reactions:
You will see from the above equations that the waste products are the ZnO and MnOOH and as these build up the internal resistance increases. The alkaline OH- (from LiOH) is both a reactant and a product so it's concentration remains constant. I tried 9V batteries which have 6 × 1.5 V cells in series. My setup and graph are shown below. I used one battery that had only a few months of use and an old one that had been in use for several years, and hence had a greater internal resistance.
I also tried new and old Eveready Gold Alkaline AA (1.5 V) batteries:
EXPT 7. Electrochemical cell and internal resistance from polarisation If you used a Daniell Cell to power some device, the cell voltage would no longer the same as the open circuit voltage of 1.10 V. This is partly because of internal resistance due to the salt bridge using up some of the cell's energy - and hence electric potential. You could try this by placing a load resistor across the electrodes and watching the voltage as time goes by. This has been detailed in previous experiments.
As the cell discharges the concentration of ions at the electrodes changes. For example, in a Daniell Cell, the [Zn2+] increases at the anode, and [Cu2+] decreases at the cathode. This is called concentration polarization. It is observed when diffusion, migration and convection are insufficient to transport the reactant to or from electrode surface at its initial rate. If the cells are agitated (by a magnetic stirrer for instance) the polarization should be reduced and the voltage be closer to the initial voltage. Heat may do the same thing by increasing the convection currents, but heat also has other effects that will complicate matters.
DISCHARGE EXPERIMENT As detailed above, a good experiment is to measure the cell voltage over time as it discharges. The higher the load resistance, R, the lower the rate of discharge. When there is no load (that is, open circuit) the rate of discharge should be very low, maybe even zero. The most appropriate quantities to plot is time, t, as the independent variable on the x-axis, and power (P = VI) in milliwatts (mW) or microwatts (µW) on the y-axis. In case you've forgotten, a watt (W) is a measure of power dissapation (P) from the cell. It is equal to 1 joule per second (1 W = 1 J/s).
RESULTS I made up a Daniell Cell using 0.10 M solutions of Zn2+ and Cu2+ (sulfates) with a saturated KNO3 salt bridge. It had an open circuit potential difference of 0.960 V. I placed a 56 Ω resistor in series with an ammeter (DMM) and placed them across the cell. I connected a voltmeter (DMM) across the resistor - or across the resistor and ammeter combined as shown. It doesn't affect the readings.
It started with a voltage of 29.8 mV (0.0298 V) and began to discharge immediately with an initial current of 0.52 mA. This worked out to a power of 15.5 µW (microwatts). The formula for power is P = V × I. Hence: P = 29.8 × 10-3 × 0.52 × 10-3 = 15.5 × 10-6 W, or 15.5 µW. The 56 Ω fixed resistor worked well but it's a matter of trial and error. If it is too low (eg 5 Ω), the current is high but the voltage goes almost to zero and changes in voltage are not measurable. If the resistance is too high (eg 500 Ω), the voltage is okay but current is too low.
Here are the rest of my results:
We can linearise the graph above by plotting the natural log (ln) of the power (y-axis) vs time elapsed (x-axis). It should be linear:
The graph is linear and has a high R2 value. This confirms the exponential nature of the discharge of a Daniell Cell.
EXPT 8. Electrochemical cells - effect of the salt bridge
In a cell that is in open circuit (no load), the nature and composition of the salt bridge has little effect on the cell voltage. I showed results at the beginning that by changing the salt bridge ions, their concentration and the sized of the pathway there is no change to the cell voltage. However, I also said that in a closed circuit (under load) changes to the salt bridge can have an effect.
NO LOAD: By 'open circuit' I mean without a load resistance across the metal electrodes. I read on one physics help-page that if you add more salt bridge strips the voltage should creep up a bit as the internal resistance of the cell would be lowered. That would make sense if a current was flowing through a load resistor - but if it was open-circuit and the only load was that of the digital voltmeter (very, very high resistance but in parallel so has no effect) the internal resistance wouldn't matter. So, its hardly worth trying. In fact, if you were going to do this for a QCAA student experiment (IA2) it wouldn't allow you to collect sufficient data for analysis. You would be trying to analyse a 'null' result. Forget about it.
UNDER LOAD: But what if the cell was under load? That way the salt bridge would be the route for the internal migration of ions. Look at it this way: a cell has an internal ohmic resistance. The salt bridge is a resistive path for the flow of ions. Ions, plus their hydration shells have low drift velocities through a medium. The salt bridge can constrain the ion current of the cell. A poor salt bridge by length, diameter, concentration, and permeability will increasingly adversely affect a cell's current and voltage as the discharge rate through the load increases. For example, under a load resistor of 56 Ω, the voltages for a single salt bridge was 33 mV and for 2 bridges it was 66 mV. So that is consistent with the statements above.
RELATIONSHIPS: The voltage of the cell (Vcell) generated by the chemical reactions is dissapated by voltage losses due to the resistance (RT) in various components of the cell. The toal resistance (RT) in this experiment will be the sum of resistances in series from the load (RL), the electrolyte solution (Rsol), the salt bridge (RSB), and the multimeter itself (RDMM). This gives us the relationship:
Load resistance (RL). This is just the resistance in ohm (Ω) of the resistor you have selected for the experiment, eg RL = 56 Ω.
DMM resistance (RDMM). When acting as a voltmeter the DMM is placed in parallel with the cell and as the DMM has a resistance of 10 MΩ it draws almost no current so has no effect on the circuit. When acting as an ammeter it is placed in series and provides almost zero resistance to the current so, it too has no effect. We can disregard the effects of the DMM on the results.
Salt bridge resistance (RSB). If you are using strips of paper soaked in a salt bridge solution you could change its resistance in three main ways:
1. Change the cross-sectional area. You could do this by changing the number of strips, or their width. Three strips will have three times the cross-sectional area as one strip. See a few paragraphs down for my experiment. 2. Change the type of ion. You could do this by changing from KNO3 to NaNO3. As K+ (220 pm* diameter) is a larger ion than Na+ (180 pm) you would expect K+ to move more slowly than Na+ and hence have a larger resistance. However, you need to look at the 'solvated' size of the ions, that is, the size of the ion with their shell of attached water molecules. Na+ solvates to three water molecules but K+ solvates to none, so in terms of solvated shell size, Na+ is much larger with a diameter of 0.5 nm (500 pm) while K+ remains at 220 pm. Thus solvated Na+ is actually twice as large as solvated K+. Now, this is really starting to get complicated. *Note: the symbol pm stands for the unit picometre which is 10-12 m. 3. Change the concentration of the salt bridge ions. It is usual to make the salt bridge out of a solution that is very conductive so it won't impede the flow of internal charge. KNO3 (Mr = 101.1 g mol-1) has a solubility of 31.6 g/100 mL at 20°C, so a saturated solution of KNO3 is 3.0 M and this works well - but you could try different dilutions. Under load you'd expect a lower cell voltage with a dilute salt bridge electrolyte (eg 0.1 M) as it offers more resistance and hence greater voltage loss. I haven't tried this.
1. Change the cross-sectional area. You could do this by changing the number of strips, or their width. Three strips will have three times the cross-sectional area as one strip. See a few paragraphs down for my experiment.
2. Change the type of ion. You could do this by changing from KNO3 to NaNO3. As K+ (220 pm* diameter) is a larger ion than Na+ (180 pm) you would expect K+ to move more slowly than Na+ and hence have a larger resistance. However, you need to look at the 'solvated' size of the ions, that is, the size of the ion with their shell of attached water molecules. Na+ solvates to three water molecules but K+ solvates to none, so in terms of solvated shell size, Na+ is much larger with a diameter of 0.5 nm (500 pm) while K+ remains at 220 pm. Thus solvated Na+ is actually twice as large as solvated K+. Now, this is really starting to get complicated. *Note: the symbol pm stands for the unit picometre which is 10-12 m.
3. Change the concentration of the salt bridge ions. It is usual to make the salt bridge out of a solution that is very conductive so it won't impede the flow of internal charge. KNO3 (Mr = 101.1 g mol-1) has a solubility of 31.6 g/100 mL at 20°C, so a saturated solution of KNO3 is 3.0 M and this works well - but you could try different dilutions. Under load you'd expect a lower cell voltage with a dilute salt bridge electrolyte (eg 0.1 M) as it offers more resistance and hence greater voltage loss. I haven't tried this.
This idea is supported by a quick test I did. I took a strip of salt bridge paper soaked in saturated KNO3 and placed it between two copper strips 10 cm apart. I measured the resistance using a DMM set to ohms (Ω). Then added a second strip, and a third, and so on. Here are my results, in a table and as a pair of graphs. The symbol n stands for the number of strips.
Summary. We can now say that the resistance of any number of salt bridge strips (in my case) is equal to 1470 × 1/n, or RSB = 1500/n to 2 significant figures. In general you would say the resistance of n salt bridges: RSB = R1SB/n, where R1SB is the resistance of 1 salt bridge.
Some typical values. In a typical experiment using 1.0 M CuSO4 and 1.0 M ZnSO4 the resistance of the solutions in a 100 mL beaker is about 28 Ω, the salt bridge is about 1500 Ω. I used a 56 Ω fixed resistor as the load. The current passes in series through these resistive components of the cell. According to Ohm's Law the current through any component is given by I = V/R so we can rearrange the equation in terms of the current I:
We can separate out terms 2 and 3 and then rearrange them:
We can now replace RT with RL + Rsol + RSB as shown earlier, but leaving out RDMM as it is insignificant when acting either as a voltmeter or an ammeter:
Replace terms with known values:
So as the number of salt bridges increases the voltage across the load as shown on the voltmeter increases.
WHY DOES THE VOLTAGE INCREASE WHEN THE CURRENT INCREASE? SHOULDN'T THE VOLTAGE GO DOWN? I'm often asked this question: "increasing the number of salt bridges increases the area for ions to move which reduces the resistance and therefore increase the current flow. According to Ohm's law, voltage is proportional to current so shouldn't the voltage loss be greater and so give a lower voltage reading?"
The answer is that resistance does decrease so the circuit current increases. But it increases through both the salt bridge AND the load resistor. Let's look at an example.
Imagine a Daniell Cell circuit which typically has an open circuit (no load) voltage of say 1.00 V in a typical student experiment. In the drawing below it has two salt bridges of 1500 Ω each. A 56 Ω load resistor is placed between the electrodes and a voltmeter is placed in parallel across the load resistor.
You can see that salt bridges are conneced in parallel between the cells. The circuit can be represented as in this schematic below, firstly with one salt bridge (on the left) and then with two salt bridges (or the right):
So you can see the voltage with 1 salt bridge is 36 mV and with two it is 69.5 mV. Double the number of salt bridges and you double the voltage. Note though, this is only for a closed circuit where a load resistor is in series with the cell. There is no point in doing this experiment unless you are using a load resistor. There would be no observable change.
EXPERIMENTAL SETUP I used 0.10 M solutions of CuSO4 and ZnSO4 with a 56 Ω load resistor. The temperature was 19.0°C. The open circuit cell potential difference was 0.93 V. I added salt bridge strips of filter paper 10 cm x 2.5 cm soaked in saturated KNO3 solution.
How to calculate percentage error. Sometimes it is easy to calculate percentage error, especially if you have a single experimental value and a single theoretical value. For example, imagine you did an experiment to determine Faraday's Constant, F, and you obtained a value of 97515 C mol-1 experimentally, whereas the accepted value is 96485 C mol-1. The percentage error, E%, is |xA− xO| /xA × 100. This gives an E% of |96485 − 97515|/96485 × 100 = 1.1%. But how do you estimate E% for two curved graph lines?
You could estimate the percentage error in the exponent: E% = |0.8705 − 0.8653|/0.8705 × 100 = 0.6%. Or you could estimate the percentage error for the constant: E% = |0.0365 − 0.0347|/0.0365 × 100 = 4.9%.
Another way is to plot a Q-Q Graph, that is, plot the experimental values (y-axis) vs the accepted values (x-axis) and calculate the equation for the linear trendline. For my data I get y = 1.06x. This means that the experimental values are 1.06 times the accepted values, or 106% of the accepted values. This can be interpreted as a percentage error of 6%.
A final way is to determione the coefficient of variability. Firstly, calculate the residual for each data point. That is, calculate the difference between accepted and observed values for each point. Then calculate the sample mean (x̄̄) and sample standard deviation (s) of the residuals (using STDEV.S in Excel). Finally, calculate the coefficient of variability, cv, for the data: cv = s/x̄̄ = 0.0033/0.0058 = 0.57. If cv < 1 then the standard deviation (s) must be small and the data sets can be considered similar (small error). In this case the cv of 0.53 < 1.0 so the data sets are similar. Hooray!
Experiments on the factors that affect the outcome of electrolysis reactions are great for student experiments. In the syllabus, there are two suggested experiments (Source: Chemistry 2025 v1.3, page 37 General Senior Syllabus, Queensland Curriculum and Assessment Authority):
For the student experiment it is better to do it quantitatively (numerical data) so that you address the criteria "thorough identification of relevant trends/patterns/relationships" and "thorough and appropriate identification of the uncertainty and limitations of evidence". A typical experiment investigating metal plating using an electrolytic cell is in the Oxford Chemistry for Queensland Units 3 & 4 Student Workbook. Here's the setup:
In essence, two copper electrodes each about 7 cm x 3 cm are weighed and placed in a 0.1 M copper sulfate solution in a beaker and connected to a 8.0 V electrical source. After 10 minutes, the electrodes are removed, dried, and reweighed. It is seen that the cathode (−) increases in mass, and the anode (+) decreases in mass by the same amount. The current must be kept constant and this is done automatically by using a constant-current DC source or manually by using a variable resistor. The voltage and temperature varies as the reaction proceeds.
QUESTION Students often ask "Why doesn't electrolysis make water decompose into hydrogen and oxygen. Why do the copper ions react to form copper instead?". The answer is fairly straightforward and is worth knowing for the external exam. If you consult the Standard Reduction Potential table you can see why. Here's an extract from the table as provided by QCAA in the Chemistry 2019 V1.3 Formula and Data book. The equations are in the same order as the QCAA table (from high negative to zero to high positive as you go down the page).
Note that the table presented in the Oxford Chemistry for Queensland Units 3 & 4 text (2019, Table 1, page 169) is in the reverse order (from + to − down the page), but the following logic is the same.
ANSWER There are three species present in the beaker in large amounts: For 50 mL of a 1M copper sulfate solution with two 7 x 3 cm strips of copper as electrodes we have: Cu (30 g, about 0.5 mol), Cu2+ (about 0.05 mol) and H2O (50 mL, about 0.3 mol). The amount of H+(aq) and OH-(aq) in 50 mL of water is extremely small (about 5 x 10-9 mol). REDUCTION REACTION: Of these three main species there are just two that can be reduced, namely Cu2+ and H2O, as shown by the equations above (Reactions I and II above), reading left to right. A measure of the ability to undergo a reduction reaction is the Eo voltage. The greater the value is in the positive direction the greater the likelihood of a reduction reaction occurring. In this case, Cu2+ is more in the positive direction (+0.34 V) than is H2O (−0.83 V). So Cu2+ will be preferentially reduced rather than H2O. OXIDATION REACTION: Of the three species present, there are two that can undergo oxidation, namely Cu and H2O (see Reactions II and III above). The oxidation reaction reads from right to left in the above equations - the reverse of the reduction reactions. A measure of the ability to undergo an oxidation reaction is the Eo voltage. The greater the Eo value is in the negative direction the greater the likelihood of an oxidation reaction occurring. In this case, Cu is more in the negative direction at +0.34 V than is H2O at +1.23 V. So, Cu will be preferentially oxidized rather than H2O. Thus, the two electrolysis reactions are:
Cathode (reduction): Cu2++ 2e-⇄ Cu(s) Anode (oxidation): Cu(s) ⇄ 2Cu2++ 2e -
WHAT METAL SOLUTIONS ARE BEST FOR ELECTROLYSIS EXPERIMENTS? Aqueous Cu2+makes a good cation for an electrolysis experiment (as shown above). Students seeking alternatives ask about zinc, aluminium and so on. But not all metal cations will work. Aluminium cannot be plated from an aqueous solution. To understand why, consider the reduction potentials. At the cathode, imagine that aluminium ions and water molecules are competing to be reduced by the electrons that have been transported to the cathode by the power source. By checking the table of reduction potentials, it can be seen that water (reduction potential of -0.83 V is much more easily reduced than aluminium ions (reduction potential of - 1.65 V). So the product at the cathode will be the reduction product of water, which is hydrogen. In fact, none of the ionic species with reduction potentials lower or more negative than -0.83 V can ever be reduced from an aqueous solution, but are ONLY ever obtained by electrolysis of a molten compound. This makes them relatively expensive even though many of them are common in the Earth's crust, due to the high energy cost of extraction. It also means that it is more vital, energy wise, to recycle metals such as aluminium, as that is far less costly than extracting more. Zinc is on the borderline, and can be extracted by electrolysis of an aqueous solution, which is the basis of electrowinning at zinc refineries. Care must be taken at the zinc refinery to ensure that hydrogen gas from the reduction of water is not produced instead of zinc. Every electron you use to produce hydrogen is an electron that is not used to produce zinc. Customers are paying for zinc, they aren't paying for hydrogen gas. There is no point converting your expensive electrons into a waste product no one will pay for. In the past, hydrogen gas was vented into the atmosphere as waste, but now refineries are capturing the gas as 'green hydrogen' to be used as fuel.
So in practice, a highish pH of the electrolyte containing zinc ions is used. These ideas are important to understand, as well as the oxidation of water at the anode at 1.23V under standard conditions, and how it affects whether oxygen, or chlorine or a mixture of the two is produced during electrolysis.
POSSIBILITIES FOR A GOOD STUDENT EXPERIMENT So copper is the best one to think about. To make a 'student experiment' out of this, you need to decide on a dependent variable and work out what factors affect it. The most obvious independent variable is mass as this can be measured accurately in any chem lab. To make it a fair test, you would choose an independent variable and then control the rest. This is the process of 'modifying' an existing experiment by a combination of refining, extending or redirecting the methodology. Here's my setup, first as a circuit diagram, and then a photo from the lab:
SOME THEORY There are many research questions in electrolysis that would lead to great student experiments. The most important concept is that of Faraday's First Law of electrolysis. It is not in the QCAA Chemistry Syllabus and so would not appear in an external exam but can be seen as the theory behind the syllabus point "state the factors that affect the products in electrolysis" (page 37). It is not necessary to describe it in a student experiment but it is useful to know if you want to design an experiment that might work, or to compare your results to theoretical values and relationships. I'll say it again - it is not mandatory but can be referred to as students wish.
Faraday's First Law of electrolysis. Michael Faraday (1833) reported that the mass (m) of elements deposited at an electrode is directly proportional to the charge (Q) in ampere seconds, or coulombs (C). Mathematically, this can be stated as: m = constant x current (in ampere) x time (in seconds), or m = constant x I x t. The constant depends on the charge on the ion of the metal reacting (eg 2 for Cu2+), and a factor that converts the amount of charge in coulomb (C) to the amount of charge in moles (mol) called Faraday's Constant F = 96500 coulomb/mol. Thus, for copper, the equation becomes:
Example: Calculate (a) the amount of copper in moles, and (b) the mass of copper in grams, that would be deposited on the cathode of an electrolytic cell if a current of 0.20 A was passed for 15 minutes. Answer: Note: this would be the same mass of copper lost from the anode.
SOME POSSIBLE EXPERIMENTAL MODIFICATIONS Here I've listed some variations that seem possible for a student experiments. The dependant variable (DV) in the following suggestions is the mass of copper deposited on the cathode or lost from the anode (should be the same). The independent variable (IV) should be either time, t, or current, I. I suggest choosing one of these two, but no other (see shaded boxes below). The rest can be done but will probably be a waste of time - as you will not be able to set up a fair test because you won't be able to control the variables that need controlling. I'll say it again: make you dependent variable the change in mass, and make your independent variable either time, or current.
An increase in concentration will lead to an increase in electrical conductivity* of the solution because of the increased abundance of mobile ions, however, the effect will be reduced at high concentrations as the ions 'associate' more due the increased crowding. Nevertheless, a larger current will be allowed to pass and this will (by virtue of Faraday's 1st Law) produce a greater mass.
* I'm using a simple definition of conductivity (or 'conductance') here as being the reciprocal of resistance (C = 1/R). Hence, the symbol for conductivity will be Ω-1.
You may note that I didn't include voltage (potential difference, or emf) as a possible independent variable. Voltage is not mentioned in Faraday's First Law and is just a means of controlling current. It would be much better to have enough voltage to get the current you want (modification No. 1 in the table above).
PARAMETERS The values of the variables in these experiments need to be in the range that is safe, and will be able to be produced in the school laboratory. We want experiments that can be carried out in a few hours at a minimal cost and be prepared by the lab staff without too many complications. But we also want to mimic as far as practicable the conditions used industrially in the electrolytic refining of copper.
Hint 1. Concentrations. The copper sulfate solutions should be in the range of 0.05 M to 0.6 M. Commercially, concentrations of 0.7 - 0.8 M are used but if you go over this you will probably get anomalous results due to 'passivation' at concentrations over 1.0 M so best to avoid. 'Passivation' is the buildup of copper ions created at the anode to the extent that they exceed the solubility product concentration needed to precipitate as solid CuSO4.5H2O. If you keep the concentration to under 0.8 M you should be okay. It does depend on the current as well so be warned. Here's a journal article with data on passivation. It's a bit heavy going but some of the data are interesting. Short answer for concentrations: keep between 0.1 and 1.0 M.
Hint 2. Volumes and containers. A suitable volume of solution is about 50 mL. That won't break the science department budget even if you do multiple trials and every group in Year 12 does the same experiment. It is possible that you could run out of 50 mL or 100 mL glass beakers if everyone is doing this experiment, and if other science classes are using beakers too. I find that the 60 mL plastic specimen jars work well. They have straight sides which makes it easy to clip the copper electrodes in place with alligator clips, and keep the strips a constant distance apart pressed to the sides of the container. These specimen jars are about 33 c in bulk (600 bottles) but more realistically about $1 each when purchased singly (eg Chemist Warehouse). At the end of the lesson the lid can be screwed back on and they won't spill - but be ready for the next lesson.
Hint 3. What thickness copper? Copper comes in various thicknesses from 0.1 mm (foil) up to about 6 mm (plate). The trouble with 0.1 mm foil is that when used as the anode the loss of copper can mean the strip gets eaten through quickly and falls apart. I'd suggest 0.5 mm thick sheet as it is a reasonable price and easy to get (if not from lab suppliers, then eBay or Amazon). I use strips about 6 cm x 2 cm and 0.5 mm thick. You don't want it much smaller as the current will be lower and you won't get as much copper lost from the anode. You need to have a reasonable amount lost otherwise the scale reading uncertainty of your electronic balance (+/- 0.01 g) will overshadow your change in mass. You want mass lost at the anode to be at least 0.1 g and even that gives a 10% scale reading uncertainty. Better to go for 0.2 g change in mass (5% uncertainty). So keep the electodes a reasonable size. You could buy 6 mm thick copper plate but that is a hassle to cut (hacksaw). Sheet 0.5 mm thick can be cut with scissors or tin snips.
Hint 4. Watch the pH. If the pH of the solution is over 4, there is a high chance of copper oxide Cu2O(s) forming on the electrodes due to a reaction between the Cu2+ and dissolved oxygen or OH- ions. If this forms and adheres to the cathode it could add to the mass of this electrode. The same is true for copper hydroxide at the anode, although this is less likely. Copper sulfate is a salt that results from the reaction of a weak base (copper hydroxide) and a strong acid (sulfuric acid). This results in an acidic solution when copper sulfate salt is dissolved in water. The pH of 0.1 M to 1.0 M CuSO4 solution made with deionized or distilled water is about 3.2 (or in the range 3.0 - 3.6) so usually there are no problems. I suggest that you check the pH of your copper sulfate solution and if it is >4, lower it by acidifying the solution before electrolysis. This will ensure that the only two copper species able to react are Cu2+(aq) and Cu(s). To acidify, add some sulfuric acid. Add a few drops of 0.1 M H2SO4 in 100 mL of CuSO4 solution should do the trick.
However, keep in mind that electrolysis is a competition between half reactions. If your voltage is too high water becomes unstable, even if you are down on the lower end of the pH scale. The hydrolysis of water can therefore compete with the plating out of or ionisation of copper on either electrode, depending on concentrations and temperature. This can cause localised pH variances, resulting in precipitation of solids, which can wreck your results. This is more likely for the lower copper sulfate concentration samples. It’s tempting to runyour electrolysis at higher voltages to reduce reaction time, but it does create some system instability. Short answer for pH: check that its below 3.5. Avoiding adding acid if you can.
Hint 5. Time. To get enough copper depositing on the cathode you need to run the electrolysis for a reasonable length of time. I would suggest 15 minutes, and if the current is great enough you should get masses between 0.05 g and 0.50 g. Most schools have available centigram electronic balances which give readings to two decimal places, for example m = 2.34 g. The rule we can use it that 'the uncertainty in a digital scale reading is equal to the smallest increment'. So, for a scale that displays to two decimal places, the uncertainty is ± 0.01 g. Thus, if the mass is only 0.05 g the uncertainty is equal to 20% of the mass (0.01/0.05 x 100 = 20%). This is very high. If the mass was 0.20 g the uncertainty of ± 0.01 g is only 5% and that should be the maximum to aim for. You may have access to a the very expensive milligram balances which weigh to three decimal places. In that case a mass like 0.050 g has a scale reading uncertainty of ± 0.001 g which is just 0.5%. No matter what type of balance you have, my advice is to make the time as long as practicible - certainly not less than 10 minutes. Remember, to do 6 trials with 3 replicates of each is 15 tests and at 10 minutes each is 3 hours of lab time. You need to get crackin'. Short answer for time: keep it between 10 - 15 minutes.
Hint 6. Current. In industry, the current used is in the range 0.2 A - 0.6 A, but because the anodes and cathodes are different sizes at different refineries, the current is specified as a 'current density' (cd) in amps per square metre. For example, refineries use current densities between 200 A m-2 and 400 A m-2. Thus, for your experiment, if you have electrodes measuring 1 cm wide and they are set up so that 4 cm is immersed in the solution, they will have a reacting surface area of 4 cm2 for each side giving a total of 8 cm2, or 8 x 10-4 m-2. For a cd of 200 A m-2 this equates to a current of 0.16 A (8 x 10-4 x 200). Short answer for current if you are making it an independent variable: keep it between 0.40 A to 1.0 A. You need to have a fairly high current to get a reasonable mass of copper deposited in 10 minutes or so. The current will be limited by the maximum voltage the power supply can produce and the concentration of the electrolyte. A regulated power supply (se below) will enable you to set the current at a desired value and it will hold that value during the experiment (provided you have a high enough voltage eg 35V). If you don't have a regulated power supply you will need a variable resistor (rheostat) in the circuit that can be adjusted during the course of the experiment to keep your current at the desired value. The voltage will not be constant and you should not try to keep it constant. It is the current that has to be kept constant.
0-30V DC 0 to 5A Regulated Power supply.
Output voltage is adjustable from 0 to 30VDC, and output current can be limited between 0 and 5A. The best part is that the output can be set to constant voltage (CV), or to constant current (CC) mode. These units are A$259 from Jaycar or A$199 each for 6+. See Jaycar Regulated Power Supply. You can also get them from Science with Mat for $219.
Hint 7. Voltage. We assume that all of the charge (electrons) that enter the cathode go towards reducing the Cu2+(aq) to Cu(s) according to the equation: Cu2+(aq) + 2e- → Cu(s). This relies on the copper ions being able to move freely from the bulk of the solution through the transition zone that surrounds the surface of the cathode to the actual copper cathode itself. This transition zone is comprised of a high concentration of ions near the surface (caused by their attraction to the free electrons of the copper cathode) which gradually decreases until you get to the concentration of the bulk solution. If the rate of transition of copper ions through the transition zone is insufficient to combine with the electrons then other cations or molecules, like water, will replace the copper cation on the surface. If the applied voltage is great enough then side reactions begin happening, and the mass of copper deposited is less than expected. This is highly dependent on the voltage applied and more likely at higher voltages. Short answer: adjust the voltage to give you the current you want. Most likely, you will want a current up to about 1.0A so a voltage in the range 8-10V usually achieves this. Do not try to maintain a constant voltage as it will vary during the experiment as the temperature of the cell increases.
Hint 8. Temperature. As electrolysis proceeds, the electric current generates heat in the solution so temperature rises. For a low current (eg 0.2 A) in a dilute solution (eg 0.1 M) the temperature rise over 15 minutes may be just 5℃, but for 1.0 A at 1.0 M it may be 20℃. This increase in temperature is something you can't avoid and can't control so you just have to work with it. The problem is that an increase in temperature increases conductivity so if you use a fixed voltage (eg 8.0 V) the current will rise during the experiment. About half of the electrical energy goes to converting Cu to Cu2+ and the other half to heating the solution.
This heating is called 'Joule heating' and is a source of energy loss in the process. Refineries try to minimize Joule heating but these thermal losses are just unavoidable. Short answer for temperature: avoid letting the cell get hotter than 80oC.
If heat losses to the environment are not minimized the rate of temperature increase falls off at higher temperature. This is consistent with Newton's Law of Cooling which says the rate of heat loss is proportional to the difference in temperature between the object and the ambient temperature of the room. This is not subject matter for senior chemistry so is best left alone. It is here to show some of the factors involved in an electrolysis cell. It was for the electrolysis of 1.00 M CuSO4 at a constant current of 1.000 A using a copper anode and cathode.
Hint 9. Maintaining a fixed voltage or fixed current. In all experiments you will need to control the current (I). If you recall Ohm's Law you'll know that voltage and current are related by the formula V = IR, which says that current is proportional to voltage when resistance is constant. You can't fix voltage and hope that current remains constant because as voltage varies so does current (and vice versa). The other problem is that temperature rises during the experiment so the relationship between V and I (the resistance) also changes. For a 'fair test' you need to have one dependent variable (eg mass) and one independent variable (eg current). You can hold some constant like the concentration of the solution, the time, the area of the plates and the distance between plates, but you can't control current and voltage at the same time. So, design your experiment with that in mind: fix the current - but don't plan to control the voltage. Short answer: fix the current and use an ammeter in the circuit to monitor it. Forget about controlling voltage. It is not relevant.
IEC power supply set to a nominal 8 V. The voltage will drift depending on the load but should stay between 7-8 V. You'll need a rheostat to keep the current at the desired value.
Hint 10. Maintaining a fixed current without a CC power supply. This is a big problem and is the major source of errors for students in these experiments. Most students don't realise that just setting a power supply at 8 V (like in the one on the leftmost photo above) will never deliver a constant current. It will rise and fall depending on the temperature and changes in concentration of the solution. What you need to do is connect it to a potentiometer used as a voltage divider. Rather than use up space describing the process here, you can download an article I prepared here.
Hint 11. Correct placement of the electrical meter in the circuit. You measure the rate of flow of charge with an ammeter. Your school will have either analog ammeters (which have a pointer and a printed scale) or with digital multimeters (DMM) which can act as a ammeter depending on the settings. The ammeter goes between the positive of the power supply and the anode.
Hint 12. Getting the data collection done in a short time. You may have just 10 hours of class time to do your prac as well as collecting data you will spend time in research and planning, analysing, interpreting and evaluating you results. If you can shorten the time in data collection then it leaves more time for the other parts. One way to do this is by running three cells simultaneously as shown in the diagram below:
The voltage will need to be three times that of a single cell to achieve the same current as for a single cell. Here's a photo of the setup:
EXPERIMENT 1: THE EFFECT OF CURRENT ON MASS. Research question: How does the mass of copper deposited vary with current when the concentration of CuSO4 and time are held constant? Expectation: According to Faraday's 1st Law, the mass deposited will be directly proportional to current when time is held constant? The concentration of CuSO4, and area of electrodes and distance apart are immaterial as they have no effect. The following graph shows the average mass of copper produced at the cathode and lost at the anode for ach of the seven trials. In each trial, a 0.30 M solution was electrolysed for 15 minutes with the current being held constant at the stated value for each trial. For example, at a current of 0.50 A, the mass of copper involved was 0.15 ± 0.01 g (0.15 g ± 6.3%). The ± 0.01 g in this case is the scale reading uncertainty for the digital scales that had a reading to two decimal places. The accuracy (or error) in the experiment can also be expressed as the percentage difference in the experimental gradient compared to the theoretical gradient. In this case it is ((0.3127 - 0.2961)/0.2961) x 100 = 5.6%. Anything under 5% would be considered quite acceptable for a high school chemistry prac (by me at least). Note that I said seven (7) trials. If you actually carry out the trial using 0.00 A (power turned off) you will get a result of 0.00 g of copper. This can be included if you actually do the experiment. It would be recorded as 0.00 ± 0.01 g because there is still scale reading uncertainty in the zero reading.
My results.
If you were wondering how the voltage changed during the experiment I have shown the results for the 0.50 A trial using below. The voltage drops because the conductivity increases as it heats up. So, to keep the current constant at 0.50 A, less and less voltage is needed as time goes by.
EXPERIMENT 2. THE EFFECT OF TIME ON MASS. Research question: How does the mass of copper deposited vary with time elapsed when the current is held constant? Expectation: According to Faraday's 1st Law, the mass deposited will be directly proportional to the time elapsed when current is held constant? The concentration of CuSO4, and area of electrodes and distance apart are immaterial as they have no effect. The following graph shows the average mass of copper produced at the cathode and lost at the anode for each of the seven trials. In each trial, a 0.30 M solution was electrolysed for seven different time intervals, with the current being held constant 0.20 for each trial. For example, at 30 minutes, the mass of copper involved was 0.12 ± 0.01 g (0.12 g ± 8.3%).
The accuracy (or error) in the experiment can also be expressed as the percentage difference in the experimental gradient compared to the theoretical gradient. In this case it is ((0.0039 - 0.0.0035)/0.0.0039) x 100 = 10.2%. Anything under 5% would be considered quite acceptable for a high school chemistry prac (by me at least) so this is not that accurate. You would need to think of ways to improve this experiment.
My results:
Students often design this experiment by keeping the voltage constant rather than the current. This wouldn't be appropriate. As the temperature change will be bigger when the time is longer, this means that the conductivity will increase with time, and hence current will increase. This means that you are not controlling the current and so the experiment is not a fair test. You'd lose marks for your design methodology. You must keep the current constant and let the voltage float (it will go lower as time progresses). For the 15 minute test I found that the voltage had dropped from 4.5 V to 4.1 V.
EXPERIMENT 3. THE EFFECT OF CONCENTRATION ON MASS (TWO APPROACHES).
Experiment 3A: Keeping current constant. Research question: How does the mass of copper deposited vary with the concentration of CuSO4 when current and time are kept constant? Expectation: According to Faraday's 1st Law, the mass deposited varies only with time elapsed and current. That is, the concentration of CuSO4 has no effect on the mass deposited when current and time are kept constant.
This experiment then will test whether the mass stays constant. It is testing the 'null hypothesis'. While this will be a fair test of Faraday's 1st Law it is not as powerful as testing a variable that will produce a change.
My results. The null hypothesis is confirmed.
Experiment 3B: Keeping voltage constant. Research Question. Students often pose the research question as: How does the mass of copper deposited vary with the concentration of CuSO4 when voltage and time are kept constant? Expectation: Students apply the logical (and correct) idea that as concentration increases the current will increase, and so the mass should increase. Hence, they state their expectation as: As concentration increases, mass will increase. Problems that students ignore: Students often do not include a voltmeter and ammeter in their electrolysis circuits and just rely on the setting of the power supply to produce a constant voltage. They assume that voltage will be constant. We know from the earlier comments that during electrolysis the cell solution heats up and the conductivity increases. This causes an increase the current and so more copper should form (according to Faraday's 1st Law, the mass deposited varies with the current if time is kept constant). This is all well and good but as the load (conductivity) increases and the current increases, the power supply often responds by lowering the voltage. And you can't tell if you don't have a voltmeter attached. The result of this is that you no longer have a fair test because one of your controlled variables (voltage) is floating. The solution is to include a voltmeter in the circuit and continually adjust the power supply to keep the voltage constant. If you have a 'constant voltage' (CV) power supply this is easy. If you have one that you just set the knob to say 8V then it may vary depending on the load. Just include a voltmeter across the anode and cathode to monitor and adjust to suit.
My results: The relationship appears to show that mass is approximately proportional to the square root of concentration. Admittedly, the exponent in the equation would be 0.5 for square root instead of 0.5931, but you get the idea. There appears to be anomalies at the higher concentrations and I put this down to polarisation (which becomes more problematic at higher concentrations). I should note that students sometimes get the opposite results to this. They get mass inversely proportional to concentration and can't work out why. I think it is because they have not controlled the voltage and just let it float.
However, this relationship is not clear as the temperature is not constant and rises more with greater currents and hence with greater conductivity. The scientific literature confirms that conductivity (conductance) increases with increasing concentration. But it is not linear. Here is the relationship taken from the literature, namely 'Electrical Conductance of Concentrated Aqueous Solutions of Copper Sulfate' by C. V. Suryanarayana and S. Alamelu, Bulletin of the Chemical Society of Japan, V32 (4), April 1959, p 333-339. Click here to download the paper.
Note that the shape is identical to our graph above. This suggests that mass would follow the same relationship with concentration because mass is proportional to current (Faraday's 1st Law) and current is proportional to conductivity (Conductivity = I/V). We now know that conductivity is proportional to [Cu2+]0.6 (from the graph below). Hence mass is proportional to [Cu2+]0.6. See if you get that.
Thius is an interesting graph (below). It shows how mass is proportional to current but I have shown the concentration I used for each of the data points. Cool huh?
Anomalous results. Students often get anomalous (inexplicable) results for their experiment and are at a loss to explain what is happening. They sometimes find that the mass of copper produced in their experiments decreases with increasing concentration instead of increasing. Here's an example from a student report:
Hint. The short answer is: control the voltage, keep concentration at or below 0.6 M.
EXPERIMENT 4: MASS AND ELECTRODE AREA. Research question: How does the mass of copper deposited vary with the area of the electrodes when the concentration of CuSO4, separation distance, voltage and time are kept constant? Expectation: The current should be proportional to the surface area as the number of mobile charge carriers (ions) will be proportional to the surface area. The mass deposited is proportional to the current, and as current will be proportional to surface area, we can hypothesise that mass deposited will be directly proportional to surface area.
My results. Note that I haven't shown mass as a function of surface area but I have shown how the current varies with surface area, and we know that mass is proportional to current. I would have expected that current is directly proportional to area but my results do not allow me to claim this. You could draw a draw a straight line through the data points and imagine that it is linear. This experiment offers lots of opportunity for a great student experiment.
EXPERIMENT 5: MASS AND SEPARATION DISTANCE OF ELECTRODES. Research question: How does the mass of copper deposited vary with the separation distance of the electrodes when the area of the electrodes, concentration of CuSO4, voltage and time are kept constant? Expectation: The current should decrease as the separation distance is increased. The mobile charge carriers (ions) will be need to move through a greater distance as the separation is increased and this will impose a greater resistance on the movement of the ions which will reducve the current. Thus, as distance increases, the current will decrease. This is an inverse relationship, possibly inverse squared. The mass deposited is proportional to the current, and as current will be inversely proportional to separation distance, we can hypothesise that mass deposited will be inversely proportional to separation distance.
Hint 1: Feathering problems. When the electrodes are close together short circuits can arise due to a process known as 'feathering'. A short circuit is where the current passes directly from cathode to anode across a bridge of copper between the two electrodes instead of through the ions in solution. The cause is quite straightforward. As Cu2+ ions are produced at the anode the solution becomes more concentrated close to the surface of the copper. Likewise, at the cathode, there is a localised decrease in concentration as Cu2+ ions are reduced to Cu. These changes in concentration result in changes in density.
At the anode the increased concentration solution is more dense than the bulk solution and sinks to the bottom of the vessel. At the cathode the decreased concentration solution is less dense than the bulk solution so rises to the top of the vessel. If the electrodes are sitting near the base of the vessel, when the higher concentration solution layer reaches the cathode, the deposition will preferentially occur at the bottom of the electrode, creating a point of deposition. This solid copper then feathers out towards the anode (the source of the higher concentration solution). Eventually this feather connects with the anode, creating a short. This results in two things. First, less material is deposited than expected due to the short. Second, this feathering material (copper) often falls off as soon as the cathode is removed from the solution and washed. This results in a lower mass than expected. You need to make sure you include the 'gangue' (fallen copper) in your deposited mass value for the cathode. The higher the concentration of the bulk solution, the faster this occurs at a given voltage.
The shorter the distance between the electrodes, the faster feathering occurs, and the higher the voltage the faster feathering occurs. Short answer: keep the electrodes several centimetres apart and not touching the bottom. Keep voltages at 8.0 V or under if possible. Keep the solution concentration at no more than 0.6 M.
My results. I didn't measure the mass involved as we know that mass is directly proportional to current. But in a student experiment you may choose to make mass your dependent variable and measure it. The trend line should be similar.
EXPERIMENT 6: MASS AND TEMPERATURE. Research question: How does the mass of copper deposited vary with the temperature of the solution when the voltage, area of the electrodes, concentration of CuSO4, time are kept constant? Expectation: The mass would increase because the conductivity would increase with temperature and so the current would increase too. This is just applying Faraday's 1st Law. The mobile charge carriers (ions) move more freely as temperature as their speed increases and the viscosity of the solution becomes less. Hint 1: This is more to do with the conductivity of the solution rather than as an electrolysis concept. It wouldn't be my first choice as a student experiment, nevertheless, feel free to have a go. The problem is that temperature will rise for any electrolysis experiment so you'll need to decide how to make it your independent variable. Hint 2: I would suggest you choose a set of conditions that give a reasonable temperature rise and keep all of that the same for each of say five trials. For example: use 0.30 M copper sulfate solution at a constant 8.00 V. Then try the experiment at various starting temperatures: eg 10℃, 20℃, 30℃, 40℃, 50℃. My results: I haven't tried this experiment but the mass will vary with starting temperature because the conductivity (and hence the current) will be greater at higher temperatures. Here's a graph of conductivity vs temperature from the literature. We know that current is directly proportional to conductivity so the relationship between mass and temperature when voltage is controlled should alos look like this. I say 'should' but can't be sure. Worth a try.
TITRATION OF TIN FROM AN ELECTROPLATED CAN
EXPT 1. Tinny taste of fruit in tin cans If you look inside an opened can of fruit you will notice that the can appears to have a bare metal surface. The surface is tin which has been electroplated over thin steel sheet, hence 'tin can'. Sometimes there is an almost invisible clear lacquer film, sometimes Bisphenol-A (BPA) - a possible carcinogen.
Tin is a fairly reactive metal and if you leave an open can of fruit for a couple of days the fruit tastes 'tinny' as the tin is oxidized by the air in the presence of food acids. The tin acts as the anode (Sn → Sn2+ + 2e-) and the underlying steel acts as a cathode (2e- + 2H+ → H2). The steel does not corrode as it is protected by the tin and because the area of iron exposed through tiny pits in the tin is small, the reaction is slow and said to be under cathodic control. If nitrates are present in the food, they will cause rapid detinning by two reactions at the cathode:
NO3- + 2e- +2H+ → NO2- + 4 H2O (slow)
NO2- + 6e- +8H+ → NH4+ + 2H2O (fast)
A tin/iron cell
Research by P. W. Board at CSIRO published in Food Technology in Australia in 1973 showed that the rate of detinning was dependent on pH and concentration of nitrate ions. The major source of high nitrate fruit at the Golden Circle Cannery in Brisbane was papaw (papaya) and if this was used in canned fruit salad and the nitrate level was high, a can with a plastic lacquer on the inside had to be used. Low nitrate pawpaw needed no such lacquer and so was preferred as it made costs cheaper. Pawpaw farmers received more money for their fruit if it was "low nitrate". I was very familiar with this process. I worked a Golden Circle Cannery as an industrial chemist in the quality control lab and wrote my MSc dissertation on nitrates in papaw canning.
Here's a good experiment: make your own tin/iron cell. You could cut up a 'tin' can into strips and stand the strips up in a beaker of dilute food acid but maybe a better way would be to construct a cell like the one shown to the left. You could vary the amount of nitrate and pH to investigate rates of detinning. How to test for tin irons? The simplest way is to do a 'iodometric' (redox) titration using a standardised solution of iodate and iodide ions as the titrant. When iodate ions (IO3-) are added to an acidic solution containing iodide ions (I-), an oxidation-reduction reaction occurs IO3- + 6H+ + 5e- → ½ I2 + 3H2O while the iodide ions are oxidised to form iodine 2 I- → I2 + 2e-. Combining these half-equations demonstrates the reaction between iodate and iodide 2 IO3- + 10 I- + 12 H+ → 6 I2 + 6 H2O. It is the iodine formed by this reaction that oxidises the Sn2+ to Sn4+ acid as the iodine is reduced to iodide ions. A starch indicator is no use in this technique as the low pH destroys it's action; you need to add an organic solvent to see the iodine clearly.
I have attached a method to download [*Vogel, 1961, p374] that will work although it is a bit more difficult than a regular titration. Colorimetric methods are difficult as the chemicals are hard to get and the methods complex: download one here [*Vogel, 1961, p 800]. The best bet if you have access to a professional lab is to have them do atomic absorption analysis for you. Your best bet may be to not use a tin can but make up a cell with tin and iron electrodes in an electrolyte of acid and nitrate. Good luck! A copy of the relevant chapter from my MSc dissertation can be downloaded by clicking here.
*Vogel, AI 1966. A Textbook of Quantitative Inorganic Analysis, Longmans, London.
EXPT 2. Detinning of tin-plated steel cans
Tin is a moderately corrosion resistant material that is widely used in tinplate for food beverage. Tinplate is light gauge, low carbon steel, coated with commercially pure tin. Worldwide, more than 90 billion food packaging cans are used per year; therefore this application is by far the largest of its diverse industrial applications. Dissolution of metallic tin from a steel can can be either undesirable or desirable; and this suggests a great EEI whereby you could look at the factors affecting the rate of detinning of steel cans by tartaric acid solution.
Undesirable: Dissolution of metallic tin from the inside of a can into the food content has a major influence on the food quality and may cause toxicological effects.
This can be caused by two major food acids: citric and tartaric acid. In an interesting paper, Professors Rabab M. El-Sherif and Waheed A. Badawy from Cairo University, Egypt, found that the presence of certain amino acids in canned fruit inhibited the dissolution of tin by tartaric acid. Their paper "Mechanism of Corrosion and Corrosion Inhibition of Tin in Aqueous Solutions Containing Tartaric Acid" was published in the International Journal of Electrochemical Science, 6 (2011), p 6469-6482. They found that the amino acids alanine, glycine, glutamic acid and histidine were inhibitors for the tin dissolution process. The bonus is that these amino acids are environmentally safe and could be added to food. You could see how the presence of different concentrations of (just one) of these amino acids inhibits detinning for a given concentration of tartaric acid (eg 1.00M tartaric) at a constant temperature over a given time. To get you started it may be helpful to know that: 0.001M glycine concentration in the tartaric acid gave a corrosion efficiency of 19% (that is 19% less dissolution than without any glycine) whereas 1.00M improved this to 67% efficiency (after 2 hours at 25°C) - with a dramatic increase in efficiency from 0.001M to 0.01M. Secondly, when the scientists compared the corrosion efficiency of different amino acids (at 1.00M for 2 hours at 25°C) they found that glycine was the best at 67%, followed by alanine at 38% and histidine at 35%.
This can be caused by two major food acids: citric and tartaric acid. In an interesting paper, Professors Rabab M. El-Sherif and Waheed A. Badawy from Cairo University, Egypt, found that the presence of certain amino acids in canned fruit inhibited the dissolution of tin by tartaric acid. Their paper "Mechanism of Corrosion and Corrosion Inhibition of Tin in Aqueous Solutions Containing Tartaric Acid" was published in the International Journal of Electrochemical Science, 6 (2011), p 6469-6482. They found that the amino acids alanine, glycine, glutamic acid and histidine were inhibitors for the tin dissolution process. The bonus is that these amino acids are environmentally safe and could be added to food.
You could see how the presence of different concentrations of (just one) of these amino acids inhibits detinning for a given concentration of tartaric acid (eg 1.00M tartaric) at a constant temperature over a given time. To get you started it may be helpful to know that: 0.001M glycine concentration in the tartaric acid gave a corrosion efficiency of 19% (that is 19% less dissolution than without any glycine) whereas 1.00M improved this to 67% efficiency (after 2 hours at 25°C) - with a dramatic increase in efficiency from 0.001M to 0.01M. Secondly, when the scientists compared the corrosion efficiency of different amino acids (at 1.00M for 2 hours at 25°C) they found that glycine was the best at 67%, followed by alanine at 38% and histidine at 35%.
Desirable: Recycling tin-coated steel cans to recover and separate the tin and steel is industrially important particularly when the price of tin is high.
Today most detinning is done by an electrolytic process whereby the steel cans are soaked in a hot caustic solution and the tin electrolytically deposited onto cathode or anode plates. An earlier method has some interesting chemistry. Research done in 1930 by Scott and Davis at UCLA (California) found that tartaric acid could be used to de-tin steel cans without dissolving any of the iron. The tin could then be recovered from the stannous tartarate solution by precipitation. Their paper was published in Industrial and Engineering Chemistry, 1930, V22 (8), p 910-911. Scott and Davis found that 3 hours of immersion in a 5% tartaric acid solution with constant aeration gave optimum detinning. You could see how the concentration of tartaric acid is related to the amount of tin removed. Question: how will you aerate the solution (does an aquarium pump give you an idea)?
HOW TO MEASURE TIN CONCENTRATIONS?
See the note in the suggestion above about colorimetric analysis of tin.
Heat of combustion High school experiments on heats of combustion usually involve burning a candle or alcohol and trapping the heat in a beaker of water. The errors are usually massive and chimneys etc are used to try to trap the heat - with little success. How could the accuracy be improved? You could explore ways and provide a theoretical reason for your trials. Alternatively, you could compare the accuracy of ΔHC values of methanol, ethanol, propanol; or even of the three C4H9OH isomers. Why might the accuracy be different? What does this tell you about intramolecular bonding? Are there any correlations with BPt?
A note of caution: teachers report that they have experienced safety issues with these burners (cracking and catching on fire, or carelessly spilt liquid igniting, and so on). Testing petrol or other highly volatile liquids seems fraught with danger but many teachers have reported that there were no problems. Strict supervision would seem necessary.
Heat of combustion of mixtures The E10 blend of fuel for cars consists of a mixture of petrol with about 10% ethanol added. It is designed to reduce the consumption of non-renewable fuels such as petrol. A good research question for an EEI would be to ask if the resultant heat of combustion of a mixture of fuels is related simply to the proportion of the fuels in the mixture and their ΔHc values (for example if Fuel A has a ΔHc of 1000 kJ/mol and Fuel B has a ΔHc of 2000 kJ/mol does a 50:50 mixture of the two have a ΔHc of 1500 kJ/mol.
Perhaps there are intermolecular interactions (eg H-bonding effects) between the components. Is there an effect with some mixtures (such as alcohols - with their H-bonding possibilities) that are not apparent with non-polar alkanes? A good suggestion from one teacher is to investigate the ΔHc of mixtures of ethanol and butan-1-ol in varying ratios (0:100, 20:80, ...80:20, 100:0). One caution: don't let the errors (which are quite extensive if you use a spirit burner as shown in the photo above) make you think there is a trend when there is not. You will need to control all other variables very carefully to keep all errors as constant as possible.
Heat of combustion of ethanol: water mixtures (and expert commentary)
Ethanol is currently being considered as a potential alternative to traditional fuels. However, ethanol offers a low return in terms of energy output per dollar invested when compared to fossil fuels. More than one-third of the cost associated with bio-ethanol production is devoted to distillation and water removal. A good EEI would be to look at the heat of combustion of ethanol-water mixtures. The research question of interest is "is hydrous ethanol a practical fuel to be used in lieu of anhydrous ethanol?" Hydrous ethanol is the chemical you have in your high school science lab: it is 95% ethanol and 5% water (E95/W5). The other type - anhydrous or absolute ethanol - is 100% ethanol and is more expensive.
In his 2012 Master of Engineering thesis from Louisiana State University, science graduate Baine Breaux investigated how water affected the combustion of ethanol. His study titled "The Effect of Elevated Water Content on Ethanol Combustion", validated up to 20% water in ethanol as a practical fuel for continuous flame applications. He said that such a fuel can be produced at a lower capital cost than pure ethanol and would provide an economic benefit despite increased volumetric consumption. The use of up to E80/W20 (that is 80% ethanol, 20% water), he said, offered a reduction in exhaust NOx concentration and a reduction in peak flame temperatures without reducing combustion efficiency or exhaust gas temperature. Baine is now Lead Development Engineer at Hiltner Combustion Systems, Washington, and gave me permission to use his graph and photos below.
Baine found that mixtures with up to 60% water can be burned but require pre-heating to 140°C (but he had a special calorimeter for this whereas you are just using a spirit burner). While ethanol has a heat of combustion of -1367 kJ/mol, for it to burn it has to be vaporised and that requires a mere +38.19 kJ/mol (= Lv). Water too has to be vaporised and that takes just a bit more (Lv = +40.66 kJ/mol). However, when you do it on a mass basis ethanol has a Lv of 846 kJ/kg whereas water has a massive Lv of +2257 kJ/kJ, almost three times as much. The poor old ethanol has to use up most of its heat of combustion just to vaporise the water. No wonder it won't ignite with too much water present. What a fantastic EEI - and think of the fantastic calculations and equations you can impress your teacher with.
I wrote to Baine asking for some advice for Senior Chemsitry students planning to do this as an EEI. His reply is as follows (7 June 2016):
1) You might not be able to burn very high water percentages; I was forcing fuel and air into a very strong mixing environment with less ambient heat loss than you will have.
2) I essentially showed lower temperatures at the same heat rate with high water content. So as you go up in water content the energy is basically transitioning from a higher temperature over a smaller mass to a lower temperature over a larger mass (water is now included). I would expect the heat transfer effectiveness into your calorimeter will change as this transition occurs, and that that heat transfer would be dominated by the temperature 'into' the calorimeter. My guess is the increased mass flow per unit fuel energy will not be captured well, if at all, and you will see a difference with different water content.
3) I don't know exactly what 'controls' the fuel feed rate in a wick. Is it the consumption rate by the flame or capillary action? Or most likely some combination of the two. You might end up supplying a similar mass or volume of fuel for your different cases, but that would give you much less fuel energy for the high water cases. If this expectation is correct then it should magnify the differences you see with different fuels. In my experiment I was able to overcome this since I was controlling fuel flow. You can at least track the fuel flow by measuring the weight of your burner+fuel. The mass-to-fuel-energy calculations ought to be good for a high schooler and add some time to your study.
Baine Breaux Lead Development Engineer Hiltner Combustion Systems 360-312-9200 Ferndale, Wa
He provided me with a revised and more concise version of his thesis. It can be downloaded here (with permission).
I took the following photos in the lab at Moreton Bay College. They show the flames for various mixtures of ethanol and water. E80/W20 means 80% ethanol, 20% water by volume. I have tried to crop the photos so they are all the same size so you can see the relative heights of the flames. The E55/W45 burnt for about 10 seconds and went out. The colours are different, perhaps indicating different temperatures.
If you are doing this investigation as an EEI you'd need to work out how to state the ΔH, as per g, or per mol. If per mol, then per mole of what - ethanol, water? And what should you control: time of burning, temperature rise? What a conundrum.
Over 90% of Australia's transportation energy is supplied by petroleum-based fuel. Enormous amounts of (non-renewable) diesel fuel is consumed annually. Alternatively, biodiesel has fuel properties similar to petro-diesel and can be used directly in a diesel engine. Biodiesel improves lubricity and reduces toxic emissions during combustion. This suggests a good EEI.
Your research question could be along the lines of which vegetable oil produces the best biodiesel in comparison to commercial biodiesel? You can make biodiesel from soybean by the following method: Weigh accurately 20.0 g of soybean oil into a round bottom flask and add a few boiling chips, 6 mL of methanol and 1.2 grams of potassium carbonate and reflux for 25 minutes (at a low intensity). Then allow to cool. Add 18 mL of 1 M acetic acid to the flask and pour it all into a separating funnel. Allow the layers of the reaction mixture to separate overnight. Drain the lower glycerol layer into a waste beaker and collect the upper layer containing biodiesel into a tared beaker. Record the mass of collected biodiesel.
Testing: place a weighed amount (about 20 g) in a clean spirit burner and set it alight (as in the two suggestions about heat of combustion above). Let the heat be absorbed into a metal can containing a small amount (say 100 g accurately weighed) of tap water. Do the usual measurements to calculate the heat of combustion per gram, and the total energy in the sample of biodiesel. There's a good article: "Soybean Oil: Powering a High School Investigation of Biodiesel" by Paul De La Rosa, Katherine A. Azurin, and Michael F. Z. Page. See attached "soybean-diesel-supporting-info".
Sugar syrup density
Sugar solutions or "syrups" are used extensively in canning, and sometimes referred to as "heavy" or "light" syrups. These terms refer to their density in grams per millilitre. Water is 1 g/mL whereas a concentrated (heavy) sugar syrup might be 1.4 g/mL. But syrups are also made up as percent by weight (%w/w).
Just how are density and percent by weight related? This might seem like a trivial question but it is important in industry because sometimes the syrup is weighed out and sometimes measure by volume. And you have to be able to move from one to the other. For instance, if you mix 100mL of sugar with 100 mL water you don’t get 200 mL of solution. The 100 mL of sugar has a mass of 160 g so you end up with 160g sugar in 260g of solution (= 62.5% w/w). The density works out to be 1.3069 g/mL.
An interesting investigation that could make a good EEI is finding the relationship between density and weight%. This has been done for sucrose and the relationship is a cubic polynomial (see my graph below). The question is: does glucose have the same relationship? The method seems quite straight-forward: weigh out accurately 50 g glucose and add 50.0g water. Mix. This is a 50%w/w solution. Tare a 10 mL measuring cylinder and add the syrup up to the 10 mL mark. Note the mass and calculate the density. I'm sure you could work out a more accurate method. Try other mixtures: eg 20% up to 70%w/w. Plot (make sure you also measure the mass of 10 mL of water (ie 0% glucose) so you can plot that point too. For extra information see the article by Karen Peterson, Department of Chemistry, San Diego State University, California "Measuring the Density of a Sugar Solution" in Journal of Chemical Education, Vol. 85 No. 8 August 2008, pp1089-1090.
Pousse Cafe (see below)
An after dinner drink called a Pousse Cafe is made from bottom to top, red grenadine, yellow chartreuse and green chartreuse. This can be simulated by coloring sugar water of different concentrations. It demonstrates how less dense liquids float on more dense liquids, if they're kept from mixing by carefully and slowing pouring them.
The most common use of nickel is for electroplating. Vast amounts of the metal are used in nickel plating and waste streams of aqueous nickel sulfate have to be dealt with. Besides being an expensive metal, nickel ions are toxic in the environment and have serious health hazards. Most commonly, the nickel (II) ions are precipitated as the hydroxide. Recovery of nickel can be right up to 100%. This suggests a great EEI, partly because the method of recovery can influence the yield.
I contacted Professor Bridget Trogden from Mercer University, Georgia, USA, who said that the mere timing of filtration was a factor affecting yield. Here's my suggestion: weigh out accurately about 1g of nickel sulfate hexahydrate (blue crystals, Mr 282.85 g/mol), dissolve in 100 mL water and add 8 mL 1.0M NaOH. You need to check the stoichiometry of this reaction to see that you have an excess of the hydroxide. Stir it and then filter immediately through a weighed filter paper. Air dry and then oven dry and weigh. I'd be doing duplicates or triplicates.
However, to assess the effect of delaying the filtering, I'd repeat the above but leave it for a week before filtering. If you have the resources, you could do another one but filter after say 4 days. You may be able to graph something here. I've attached the method from Professor Trogden's article in J Chem Ed, V88(2) Feb 2011, pp192-194 here.
She says that there are risks with using nickel compounds but should be safe for high school. The experiment contains no chemical hazards. Nickel compounds are considered suspected cancer agents via inhalation, but exposure is not expected in this experiment. Additionally, nickel hydroxide substances do not cause chemical burns or other problems that harsher hydroxides can. A 2005 J. Chem. Educ. Letter addresses these concerns and points out appropriate literature [Bentley, Anne K. et al. J. Chem. Educ. 2005, 82, 1775]. You'd do a risk assessment anyway. If it seems too dangerous you could try copper sulfate. Nickel sulfate hexahydrate (Sigma Aldrich) costs about A$37 for 100g.
The increase in ocean acidity since preindustrial times may have deleterious consequences for marine organisms, particularly those with calcium carbonate structures. The intergovernmental Panel on Climate Change (IPCC 2013) states that "carbon dioxide concentrations have increased by 40% since pre-industrial times, primarily from fossil fuel emissions and secondarily from net land use change emissions. The ocean has absorbed about 30% of the emitted anthropogenic carbon dioxide, causing ocean acidification".1
A good EEI would be to take some calcite to represent the calcareous organisms. Calcite is the most common form of the mineral whereas marble is the metamorphic form. Really, I guess you could use either. To a weighed sample you could add some acidic buffer solution of various pHs and let it react for a set time (maybe 30 minutes). Dry and reweigh. If the pH drops during the reaction add more buffer. What does the graph look like? Is pH that critical? Perhaps you should do triplicates. What about temperature (ocean warming): you could try a change in temperature as a separate variable. I think you may be shocked. There is an interesting article titled "Laboratory Experiment Investigating the Impact of Ocean Acidification on Calcareous Organisms" by Alokya P. Perera and A. Bopegedera in the J. Chem. Educ. 2014, 91, 1951−1953.
1. http://www.climate2013.org/images/uploads/WGI_AR5_SPM_final.pdf (accessed October 2015).
Corrosion Corrosion happens all around us - our cars rust, bridges and other steel structures fail, and we spend billions of dollars each year in replacement and maintenance costs as a result. There are a number of methods used to minimize or prevent corrosion, which include alloying, metallic coating, organic coating, use of inhibitors, and anodic or cathodic protection. Corrosion is one of the more popular topics in Queensland schools for an EEI as we have a warm, humid climate and the bulk of the population lives along the coast.
Iron is the most abundant metal on earth and has been a boon to the building industry since the Iron Age. However, as it is susceptible to corrosion or rusting, structures made of iron, such as bridges and ships, need to be regularly monitored for rusting. If not, the damage caused by rusting can be very expensive to fix and, perhaps, hazardous. This is a particular problem in the shipping industry where the moist, salty conditions are ideal for accelerating the rusting process. For shipping and many other uses, iron is converted to one of its alloys, carbon steel, to make it stronger and less susceptible to corrosion. Salinity is only one of many factors that will contribute to the nature and extent of iron and steel corrosion observed at shipwrecks. Others factors, such as the concentration of dissolved oxygen, pH, temperature of the water, among others, may have a significant bearing on the corrosion of a particular wreck. That is why shipwrecks at different ocean depths and latitudes may vary in the nature and the extent of corrosion.
Good EEIs often are in the context of shipwrecks. If so, it is not enough to merely put steel nails in different solutions and look at the loss of iron. You should be looking at the environments that ships can be found in and considering how you can simulate corrosion on a speeded up scale. Also important is what you will use for the metal: steel may be okay - but what alloy is it? Is pure iron any use - with no carbon to act as active sites for corrosion? Teachers who have been to Australian Corrosion Association conferences say that their website has useful information. As far as the best way to measure the amount of rusting, you may like to contemplate the advice given by Chemistry teacher Daniel Bischa from Pioneer State High School, Mackay, Queensland. Download his comments here.
If you leave steel out in the air it will rust as the iron reacts with oxygen and water in the air. If you exclude water the iron will still react with molecular oxygen thus: 4Fe(s) + 3O2 (g) → 2Fe2O3 (s)
As the reaction proceeds in a sealed container (eg a test tube), the oxygen will be consumed and so the pressure inside the tube will decrease. If you use an oxygen pressure sensor (eg Vernier) connected to a laboratory data collector (LabQuest, Datamate, and so on) you could plot a 'percent of oxygen' versus time graph. From this you could determine the order of the reaction, or more simply, determine what factors affect the rate of reaction (temperature is an obvious one). The rate can be determined from the graph you plot.
As a reminder, let's imagine it could be 1st, 2nd, or 3rd order:
So, you'd plot all three forms of the graph to try and linearise the relationship, and if one gives you a straight line then bingo - that's it.
Corrosion of iron using spectrophotometry
Corrosion of iron in salt water is a popular context for an EEI. It is topical, it is of great economic importance, and it makes a great experiment. A common way of determining the corrosion rate is to measure the weight loss of an iron nail after a wee in salt water. However, to get reliable results, a balance with a precision of 0.01 mg is required since the weight loss is quite small over the week. And who has a spare $10000 to buy such a balance? But here’s a good spectrophotometric method that can be done with some simple chemicals and a phone with a colour analyser app.
In essence, sandpaper a nail with a wet 1200-grit sandpaper and degreasing it with a dip in acetone. Air dry it and immerse in 100 mL of a solution containing 3.5% NaCl, 1% ascorbic acid, and 0.25% 1,10-phenanthroline and begin timing. The color of the solution turns orange as time goes by. This is because the 1,10-phenanthroline,4,5 molecule reacts with iron to form an orange tris(1,10-phenanthroline)iron(II) complex ion. The concentration of this ion is directly dependent upon iron concentration.
Fe2+(aq) 3 phen → [Fe(phen)]32+.
The phenanthroline presents no chemical hazards and is available in a 5g bottle from Chem-Supply (Australia).
You can carry out the reaction on a magnetic stirrer if the iron nail sample is magnetic, and itself will function as a stir bar. Take aliquots (samples) of the solution every 5 minutes, transfer into a test tube or plastic cuvette, and capture the image with a mobile phone. I tried both Color Picker (for iOS on an iPad or iPhone), and Color Grab for Android. They both worked well. Of course you’ll have to make up a set of standard solutions of iron (II) ions with 1,10-phenanthroline added and measure their spectra.
A good source of Fe(II) is ammonium iron (II) sulfate hexahydrate and a standard solution of about 1 g/L is good. Then make up a range from 0.1 to 8 mg/L in 25 mL volumetric flasks for the standard curve. If you use the RGB values from the phone, you can calculate the absorbance. In essence, you subtract the blue value from the green value for the sample. You divide this by the B minus G values for the water blank. Then take the negative log (10) of the result. The value used by scientists is somewhat different (J. Chem. Educ. 2015, 92, 1696−1699, Supplementary Material). For example: A = -log((0.76Gs+0.5Bs)/(0.76Gb+0.5Bb) where Gs is the Green reading for the sample and Gb is the green reading for the blank. Likewise for blue (B). The value for R is not used as it is assumed to be zero
My thanks to Edgar Moraes, Mario Confessor and Luiz Gasparotto, Federal University of Rio Grande do Norte, Brazil.
pH and photosynthesis Oxygen is evolved during photosynthesis but the conditions for maximum reaction rate are intriguing. It can be affected by many things, including: sunlight - its intensity and wavelength, temperature, CO2 and O2 availability, water (which closes stomata and restricts CO2), and any factor that influences the production of chlorophyll, enzymes, or the energy carriers ATP and NADPH, such as pH and Mg2+ availability. You could test the effect of pH and temperature. It sure won't be linear but how well your prediction (hypothesis) and results agree will be interesting. You could also try light intensity. If you don't have a "luxmeter" to measure intensity you could take advantage of the fact that as you double the distance of the light source to the plant, the intensity is quartered (but you'd have to cut out daylight). There are a lot of variables to control and complex biochemical reactions to examine.
Chlorine loss in a swimming pool - due to sunlight intensity Home swimming pools are usually sanitized with chlorine-based compounds such as calcium hypochlorite, Ca(OCl)2 or sodium hypochlorite NaOCl, which produce the hypochlorite ion HClO- when dissolved in the pool water. Chlorine in a pool can get consumed in many different ways, but the most common is from sunlight and aeration which convert chlorine in an oxidation state of +1 into chloride ion in an oxidation state of -1. Reports suggest that in strong sunlight, up to half of the HOCl is destroyed within 17 min. A good EEI would be to make up some pool water and add a measured amount of either calcium hypochlorite or sodium hypochlorite and measure the rate of consumption of free chlorine in pool water when exposed to sunlight. As a second IV you could look at the rate of loss at different pHs.
The standard method for determining free chlorine is to measure the amount of oxidant by its ability to liberate iodine from acidified iodide solution. Take a chlorine-containing water sample, add an excess of KI solution to liberate free iodine which produces an indigo-blue colour formed with a fresh starch indicator. Find the amount of this iodine released by back titration with sodium thiosulfate. Click here to see a good method. The problem with the iodometric (iodine titration) method is that it takes a long time for students to collect data. Janet Grice suggests Doug De La Matter's Methyl Orange method. Her Yr 12 Pool Chemistry handout is also available. And I've attached an article from Chem Matters supplied by Janet Grice. As another IV you could look at amounts of aeration by bubbling air through it. Note the warning below!
Chlorine loss in swimming pool water - dependence on colour Chlorine loss from pool water is known to be due to the action of sunlight (see text above). However, it is possible that the breakdown of chlorine is greater for different wavelengths of light than others. For example, does it breakdown as quickly under red light as under blue light? It would be an interesting EEI to see which colour/s have the greatest effect. You could make up some pool water with a known amount of chlorine (using Ca(OCl)2 or NaOCl), place in a stoppered test-tube (why stoppered?) and wrap in a single layer of cellophane. You should be able to design the rest of the method yourself but you'd need several colours of cellophane and to measure the free Cl at several intervals of time (see experiment above for titration suggestions).
Your problem will be to ensure the same intensity of light gets through to the solution (yellow may not absorb as much as blue for instance). The image below shows the wavelengths of light most transmitted (passed) by each type of cellophane; this is called their "λTmax" (lambda T max), that is, the wavelength most transmitted. I did this on a spectrometer at Moreton Bay College but you could run them again if you can get access to a spectrometer. You would also need to know what % transmission occurs for each colour; I didn't do that. As a second IV you could try thickness: one layer, two layers etc of cellophane to see if the response is linear. Have fun!
Chlorine loss in swimming pool water - the role of cyanuric acid stabilizer. The biggest problem with chlorine as a sanitiser in swimming pools is that it breaks down and dissipates very easily under the sun's radiation. This can be fixed by adding cyanuric acid. Cyanuric acid (1,3,5-triazine-2,4,6-triol) is used as a "stabilizer" for chlorine in swimming pools and stops it breaking down so quickly in sunlight. On a bright sunny day, nearly all of the chlorine in a pool can be lost in less than two hours unless a stabilizer (like cyanuric acid) is present. The addition of about 30 mg/L (ppm) cyanuric acid to swimming pool water reduces destruction of the free chlorine by sunlight. In the stabilization process, a portion of the chlorine residual is temporarily bonded to the cyanuric acid molecule which protects the chlorine from the destructive effects of sunlight. The nature of this bond is such that the chlorine continues to be released as long as a demand exists.
The ideal level of cyanuric acid is 30-80 mg/L but no more than 100 mg/L (100 ppm) as a maximum. An interesting EEI would be to assess the ability of cyanuric acid to prevent the degradation of chlorine in water when exposed to sunlight. Perhaps you could add solutions of varying concentrations of cyanuric acid (eg 0 to 100 mg/L) to some water which has chlorine present (maybe 10 mg/L) and put it in the sun (or fluorescent light) for so many hours. At the end, you could measure the concentration of chlorine and see if there is a relationship between loss of Cl and concentration of cyanuric acid. Secondly, you could take this EEI further (but this will be harder): perhaps of the cyanuric acid breaks down too as it tries to prevent chlorine loss. It is not supposed to but you could check.
The test for cyanuric acid is a reaction with a melamine solution which forms a fine, insoluble, white precipitate (melamine cyanurate) that causes the water to cloud in proportion to the amount of cyanuric acid in it. For a school chemistry EEI you could buy a cyanuric acid test kit from a pool shop (about $30). The kits have a range of 20-100 mg/L but in 10 mg/L increments - which is not that accurate. There are two main types, one (called "the disapppearing dot") where you mix an equal volume of the test solution (see photo below, right) with your sample and add it to a graduated tube until you can no longer see a black dot on the bottom. The level of the liquid when this happens gives a reading on the side of the tube in mg/L of cyanuric acid.
The second method is where you add your water to a tube (see photo below, centre) and add a melamine tablet and crush it. The solution will go cloudy and you raise the black dot on the bottom until you can just see it. A scale is on the lifter is graduated in mg/L. To be more accurate, you could prepare a set of standard cyanuric acid solutions and measure their turbidity in a spectrometer (λ max = 420 nm) after the test solution or tablet is added from the kit. This will allow you to prepare a standard curve from which your experimental solutions can be compared. A method for this was published in the Water Research journal. Click here to download an extract.
Effect of copper on the growth of algae (may be more suited to a student doing Biology). The last thing you want in your swimming pool is algae - the green plant that grows on the walls and bottom of the pool. There are several ways to control it: keeping the sanitizer (chlorine) levels correct helps but often a copper-based algicide (algae killer) is used. The copper ion (Cu2+) is a very effective algicide to both kill and prevent algae formation. Swimming pool companies say that about 0.03 to 1.0 mg/L (0.03 to 1.0ppm) of free copper ion must be present to be effective and safe. The word "free" is used because "bound" copper (copper is tied up in an insoluble form) is not available to work as an as algicide. For non-biological systems (where no living plant or animal is present) a continuous level of 1.0 ppm is enough to assure effective algae control; more is superfluous and may damage surfaces and equipment.
The toxicity of copper to algae has been the subject of a number of studies over the past 40 years because of its widespread use for the control of algae in natural waters. This suggests a good EEI. You could try growing algae in solutions with different copper ion concentrations from say 0 to 1 ppm. One problem you will have to sort out is how to measure the amount of algae in the samples. Perhaps it can be done using a spectrometer, or by measuring the depth at which you can just see a black cross appear/disappear (like in a simple nephelometer tube they use in Biology, or like the Secchi Disk method for turbidity in natural waters). Safety note: copper is a heavy metal ion and is considered hazardous. It is important that you become aware of the risks. Care should be used when handling this product.
An interesting study by Drs Jenny Stauber and Mark Florence from CSIRO's, Division of Energy Chemistry, Lucas Heights Research Laboratories, Sydney, Australia found that copper ions depressed both cell division and photosynthesis in many species of algae notably the common freshwater green alga, "Chlorella" (Chlorella pyrenoidosa). Reference: J. Stauber and T. Florence, 'Mechanism of toxicity of ionic copper and copper complexes to algae', Marine Biology 94, 511-519 (1987).
In their experiment they maintained Chlorella pyrenoidosa in MBL medium on a 12 hour light: 12 hour dark cycle (Philips 40 W fluorescent tube, white, 6500 K - see photo below) at 21°C. They found that a Cu2+ concentration of 7.9 x 10-7M (5 x 10-5 g/L, equal to 0.05 mg/L or 0.05 ppm) gave a 50% reduction in growth. Click here to see what the MBL medium consists of (this may be too complicated for high school EEI). One question you need to sort out is how to measure algae growth (perhaps measure the absorbance in a spectrometer).
If you don't do Senior Chemistry you may need to brush up on your formulas for amounts and concentration. The copper sulfate your school lab has is most probably copper sulfate pentahydrate (CuSO4•5H2O). It has a molar mass of 249.5 g/mol. Copper itself has a molar mass of 63.5 g/mol. Thus, to make a 1000 mg/L Cu2+ solution (1000 ppm) you would have to weigh out 1000 x 249.5 ÷ 63.5 g of CuSO4•5H2O per litre of distilled water (3.929 g/L). Make sure you use distilled water as tap water will go cloudy. You can then do serial dilutions (1:10) to reduce this to 100, 10, 1, 0.1 ppm Cu2+, and from there you can make the solutions you want.
Precipitation of copper carbonate in swimming pools To kill algae in a swimming pool either ionic copper (in the form of copper sulfate) or chelated copper can be used (see above). Pool manuals and pool chemical suppliers say that the problem with using the ionic form - copper sulfate pentahydrate, CuSO4•5H2O - as the algicide is that it doesn't last long in pool water. Pool water has carbonate ions (CO32-) present from the addition of sodium carbonate or sodium bicarbonate as a buffer against pH changes. The carbonate ions react with the added copper sulfate to form a precipitate of copper carbonate: Cu2+(aq) + CO32-(aq) → CuCO3(s). So it would appear that any copper ions added to pool water would immediately be precipitated as the carbonate and thus not available to kill algae.
But pool chemical suppliers say that the copper ions work for several hours which is enough time to bust open the algae's cells and kill them. This suggests a fascinating EEI. You could look at the rate of precipitation of copper carbonate in aqueous solution (pool water). The solubility product (KSP) of copper carbonate at 25°C is 1.4 x 10-10. The equation KSP = [Cu2+(aq)] [CO32-(aq)] = 1.4 x 10-10 means that if the ionic product of [Cu2+(aq)] and [CO32-(aq)] is greater than 1.4 x 10-10 precipitation will occur.
Pool water typically has a carbonate ion concentration of 100 ppm (mg/L) expressed as CaCO3. Using relative molar masses this means the actual carbonate ion concentration is 60/100x100 = 60 mg/L or 0.0001 M. You do the maths! It is also recommended that the copper ion concentration in pools be about 0.05 ppm (mg/L). This is about 0.0001 M. The ionic product is 1 x 10-8 which is greater than the KSP so a precipitate should form. However if both the copper and carbonate concentrations are 0.00001 M each the Trial Product is 1 x 10-10 which is less than KSP so no precipitate should form. You could try various (equal) concentrations of Cu2+ and CO32- and examine the turbidity of the resulting solutions using a spectrometer.
I would suggest a wavelength of 400 nm (although Balch recommends 560 nm for turbidity (R. T. Balch, Measurement of Turbidity with a Spectrophotometer, Ind & Eng Chem Anal Ed V 3, no. 2, p124-5). I got higher absorbances at 320 nm (UV) and 820 nm (near IR) but these may have been artefacts of the instrument (the plastic in the cuvettes absorbs strongly in the UV). Most importantly, you could see how the turbidity varies with time (perhaps every hour or every day) as the pool chemists suggest. If you don't have a spectrometer you could look at settling rates of visible precipitates (> 0.0025 M solutions) as a function of concentration, temperature or pH.
Getting PAM to clarify dirty water You may have seen the ad for the World Vision charity where the little African girl is holding a plastic bottle full of dirty water for drinking. Communities like hers benefit from clean drinking water and one way to achieve this is through sanitising (with chlorine) and clarifying - using a "coagulant" that causes the suspended particles to coagulate (come together) and settle to the bottom ("flocculation") and are big enough to filter out.
Two coagulants/flocculants commonly used for water are alum (aluminium sulfate Al(SO4)3•nH2O, where "n" is usually 14 or 18). This is an inorganic flocculant and is discussed in the next suggested EEI further down this page. The other type of flocculant is the organic polymer type and the most common of these are called cationic polyelectrolytes. Two examples are polymer polyacrylamide (PAM) and poly di-methyl di-alloyl ammonium chloride (PolyDADMAC).
The process is simple: the coagulant is added to water mixture and is then slowly stirred in a process known as flocculation. This water churning induces particles to collide and clump together into larger lumps, or "flocs." The coagulant works by creating a chemical reaction and eliminating the charges (negative or positive) that cause particles to repel each other. The process requires chemical knowledge of source water characteristics to ensure that an effective coagulant mix is employed.
Improper coagulants make these treatment methods ineffective. These polyelectrolytes are not only used for drinking water; they are used in industry for such applications as clarifying paper mill wastes and dewatering primary and secondary activated sludges. This suggests a good EEI.
You could make up some "dirty" water by adding clay - not too much, maybe 1 g per 5 litres - and adding an organic polymer flocculant such as PolyDADMAC or PAM - and giving it a good stir. I'd suggest using some terra cotta clay from the Art Department of your school as it is a nice red brown colour and the floc easy to see.
Don't let them give you "paper clay" as it has ground up paper that stuffs things up. You'd need at least five different amounts and probably duplicates or triplicates (trials) of each. Your problem is also to control the variables: how long to stir for, how fast to stir, how long to allow settling, what to measure (height of floc, turbidity of "supernatant" liquid (clear liquid above the floc). Other variables to try: temperature, pH, salinity. Instead of clay, you could use CaCO3, BaSO4 or limewater Ca(OH)2. What might be a good Research Question? It is not much good just saying "do organic polymer flocculants clarify dirty water?" - because you know they do. Perhaps "Is PAM or PolyDADMAC better at clarifying muddy water?"
At some stage you need to develop an hypothesis. Perhaps there is an optimum amount of flocculant that can be used (too much or too little doesn't work at well). You'd also need some theory to support your hypothesis. Where to get the flocculants? The cationic PolyDADMAC is available as Focus Brand "Water Polish" or many other proprietary names. Polyacrylamide (PAM) is also available from pool shops with brand names like Aquatic Element's Aquatic Clear Advantage, Premium Quality's Ultimate Clarifier, Bioguard's Polysheen Plus, or PowerFloc. Note: don't get mislead by the internet: polyacrylamide is also available from the gardening section of a hardware shop as "water retaining crystals" - brand names like Hortico (see below); and surprisingly also available from shops that sell disposable nappies (eg Huggies, Pampers) which have the polyacrylamide as the water absorbent (rip one open).
However, these forms of polyacrylamide are NOT suitable as they are not activated and don't work - I've tried it. Stick to the pool shop product. One last warning: I had terrible trouble getting PAM to coagulate muddy terra cotta water, whereas PolyDADMAC worked quickly. Why is that? Maybe the charges on the clay ions are not neutralised by PAM. Hmmm, a good EEI.
Clarification of water with alum Cloudy water for domestic water supplies is commonly treated with alum (Al(SO4)3•18H2O). The name 'alum' is a bit confusing as there is also a double sulfate of potassium and aluminium with the formula KAl(SO4)2·12H2O) commonly called 'alum'. The 'alum' used as a coagulant is the first one: aluminium sulfate - but be aware that there are several types of aluminium sulfate, each with different amounts of water of crystallization. The most common in schools is the one with •18H2O, sometime called octadecahydrate (Mr = 666.4). The other common one is •14H2O.
The reason I mention this is because they will have different molar masses and this will be important when weighing it out. Alum acts as a coagulant, which binds together very fine suspended particles into larger particles that can be removed by settling and filtration. In this way, objectionable color and turbidity (cloudiness), as well as the aluminum itself, can be removed from the drinking water. By the addition of a small amount of alum to water, it can be filtered through ordinary paper without difficulty, and yields a brilliantly clear filtrate, in which there is no trace of suspended matter.
If it believed that alum not only clarifies a water, but also removes disease germs and ptomaines, so its use is of incalculable value to society. A good EEI would be to make up a sample of water with suspended clayey matter and then filter it through the best filter paper you have at school. To the (still) cloudy filtrate you could add alum solution (about 20 to 1000 mg/L) to see if it settles the clay and enables you to filter the solids out (weighed filter paper). Here are some ideas for your hypothesis: try different amounts of alum (there is an optimum amount - too much alum will actually impede the coagulation/flocculation process.
Try different acidity/alkalinity as the pH is a very important parameter in water treatment, especially for effective coagulation. Each coagulant has a narrow optimum operating pH range. For example, alum tends to work best at a dosed-water pH of 5.8-6.5). Aluminium sulfate should be readily available at school - if not then go to the pool shop. Remember to include the •18H2O (or whatever) in the formula when working out the molar mass. A great EEI with great social importance.
Reaction rate by the scattering of light #1 Reaction rate experiments make terrific EEIs as you can collect reliable data quickly, draw graphs and talk about all of the problems. Here's one of my favourites. It is one you have probably done in Year 10 but in a rather inaccurate way. It is the reaction between hydrochloric acid and a sodium thiosulfate solution whereby yellow sulfur precipitates out of a colourless solution. The reaction is this:
HCl + sodium thiosulfate → sodium chloride + sulfur dioxide + sulfur + water.
2HCl(aq) + Na2S2O3(aq) → 2NaCl(aq) + SO2(g) + S(s) + H2O(l) Equation 1
This reaction rate depends on the concentration of the two reactants and the temperature. Because the sulfur particles are so fine they form a cloudy whitish colloid. The concentration of the sulfur can be measured by the 'cloudiness' of the colloid mixture. The rate of this reaction can be measured by looking at the rate at which the product solid sulfur S(s) is formed. You may have carried out this reaction in Year 10 during the chemical reactions unit. It is usually carried out in a flask placed on a piece of white paper with a black cross on it - and you time how long it takes the cross to disappear. It is an old favourite. In effect, you are making use of the Tyndall effect (or Tyndall scattering), whereby light is scattered by particles in a colloid [see note below].
I tried it again using a light meter. I made up a cell from four microscope slides epoxy glued together along their long edges to make a hollow box, and then glued another slide on the end (see photo above). I put a solution of 0.8 g of sodium thiosulfate in 50 mL water into my cell and shone a beam of light from a LED torch through the mixture and measured the intensity (transmittance) of the beam using a light sensor. I also tried a laser pointer (green, λ = 566 nm) and that worked well too.
To measure the transmitted light through the base of the cell, you could use a digital light meter that comes with the various laboratory data kits (eg LabQuest, DataMate, Spark). I tried an Arduino with a light dependent resistor (LDR) and it worked well. I then added one drop (0.05 mL) of 1M HCl to the cell, quickly stirred it and started a stopwatch. As time went by the solution became more and more cloudy and the transmittance decreased. I 'normalised' this value by expressing the transmittance as a fraction of the initial transmittance (Tn = T/Ti).
The reaction is said to be first order with respect to sodium thiosulfate concentration (rate ∝ [A]), where [A] is the concentration of the sodium thiosulfate. However, both the overall chemical equation and the mechanism for the decomposition of sodium thiosulfate are more complex than suggested by Equation 1 above. The reaction is acid-catalyzed, which means that the acid concentration must have some bearing on the rate in terms of producing an equilibrium concentration of HS2O3– ions. The HS2O3– ion is a reactive intermediate, reacting further with additional S2O32– ions to produce polymeric ions containing multiple S atoms. When the chain of S atoms in a polymeric ion becomes long enough, it "closes" in on itself to form a ring of elemental sulfur (S8). That is complicated and the order is anyone's guess. Here are the steps:
S2O32– + H+ → HS2O3– H—S—SO3– + nS2O32– → H—S—(S)n—SO3– + nSO32– H—S—Sn—SO3– → H+ + –S—Sn—SO3– –S—S7—SO3– → S8 + SO32–
NOTE: For a dispersion of particles to qualify for the Rayleigh formula, the particle sizes need to be below roughly 40 nanometres, and the particles may be individual molecules. Colloidal particles are bigger, and are in the rough vicinity of the size of a wavelength of light. Tyndall scattering, i.e. colloidal particle scattering, is much more intense than Rayleigh scattering due to the bigger particle sizes involved.
Reaction rate by the scattering of light #2 Further to the above discussion, you could also look at the light scattered at right angles to the path of the beam. As shown above when light is passed through a suspension of sulfur it is attenuated. That is, the transmitted light is not as intense as the incident light. This happens for small colloidal particles in suspension such as sulfur. The process shown in the EEI above is called 'turbidimetry': it is the measurement of the degree of attenuation of a radiant beam incident on particles suspended in a medium, the measurement being made in the directly transmitted beam.
But light is also scattered in all directions as it passes through the suspension. You can also measure the 'scattered light' by a technique called 'nephelometry': the measurement of the light scattered by suspended particles, the measurement usually being made perpendicularly to the incident beam. Turbidimetry or nephelometry may be useful for the measurement of precipitates formed by the interaction of very dilute solutions of reagents (great for this Chemistry EEI), or other particulate matter, such as suspensions of bacterial cells (Biology EEI).
A fabulous chemistry EEI would be to examine the light scattered (by Tyndall scattering) through the sides of the glass cell. You could even do it in a beaker. If you look at the photo in the EEI above (#1) you will see that I also sticky taped a light dependent resistor (LDR) on the side of a cell, as well as the one underneath it. I connected the wires to the analog inputs of an Arduino board (each in series with a 1000 ohm resistor) and used a 'sketch' that read the outputs across the LDRs. It is very simple and worked well. The circuit diagram and the sketch is here if you want it.
In the graph below I have plotted the scattered intensity against time. The scattered light will start at a low number and then increase as more sulfur is precipitated. It is like the inverse of the transmitted light graph (although there is no mathematical relationship between transmission and scattering).
Reaction rate by the scattering of light #3 - effect of concentration The kinetics of sodium thiosulfate and acid reaction (above) may he studied in further detail by recording different times required for solutions of different concentrations to attain the same degree of turbidity (that is, the same scattered intensity reading as measured by a light meter). You may recall from your Year 10 science that the time is approximately inversely proportional to the concentration. This can be established mathematically for a first order reaction. I thought I'd try it experimentally.
If you are using a light meter then just record the the scattered intensity as a function of time (as above) as you try out different concentrations. I made up four solutions of sodium thiosulfate in 50 mL water (0.2g, 0.4g, 0.6g and 1.0g), added acid and measured scattered intensity. Here are my four graphs (left):
This suggests a great EEI. The hypotheses are obvious from the above, but it is the measure of light intensity that needs care. Judging brightness with your eye is difficult and not that accurate. You may have a light intensity probe available with some of the datalogger kits. If not, you could use a light dependent resistor (LDR) such as the ORP-12 for which there are calibration curves of intensity vs resistance (see below). The key think is to get absolute darkness as you don't want ambient light (room or sun) getting in.
I made a colorimeter by using a length of grey plastic tubing (eg electrical conduit) with an inside diameter big enough to fit a test tube (eg 20mm). I cut a window in the side the diameter of the LDR and glued the LDR in place and wrapped black tape around it to stop light getting in. You'd be surprised how light makes its way in through the glue. I connected the two leads to a multimeter and glued an endcap on the bottom (you can buy them to fit or just use a metal screw-cap from a wine bottle.
Reaction Rate and Surface Area - I Controlling reaction rates is one of the great challenges facing scientists and engineers in modern day life. You put food in the refrigerator to slow down decay, and you use hot water to wash up as fat reacts faster with detergent in hot water. Temperature is one way - but so is the control of surface area: fine sugar dissolves faster than coarse sugar; sawdust burns quicker than a lump of the same wood; drugs are made with different particle sizes to control the speed of release into the blood stream. This seems like the basis of some great EEIs.
In Year 8 you probably did a science experiment with Alka Seltzer tablets to see what factors affected how fast the tablets dissolved. You would have looked at temperature (tried hot and cold water), and surface area (whole tablets versus crushed up ones). However, for surface area you would not have done it quantitatively (numerically, by calculating the surface area using a ruler). This suggests a great EEI but if you plan to do it with Alka Seltzer tablets the chances of getting an "A" will be greatly limited by how well you can control the variables. If you were to try it as an EEI you could try break up tablets into 2, 4, 6, 8 pieces and measure how long they take to dissolve and react. You could measure the sides of the chunks with a Vernier calliper and calculate the surface areas. I get a diameter of 25.63 mm, thickness of 4.30 mm and a full surface area of 1305 mm2. When split in halves I get a SA of 1461 mm2.
However, the reaction time for a whole tablet (at 23.5°C) is about 63 s, halves 54 s, quarters 51 s, eighths 43 s. A completely crushed tablet - a powder whose surface area is enormous - takes about 23 s. So it is not linear; that is, the reaction rate doesn't vary directly with surface area.
What's the surface area of a powdered tablet? It is impossible to measure this directly in a high school laboratory. However, based on an estimation of how fine the powder is ground up (in a mortar, with a pestle) you can state an approximate "Specific Surface Area" (SSA). This is expressed in square metres per gram. For example, coarse sand (0.5 - 1.0 mm diameter) has a SSA of 0.01 m2/g; fine sand 0.06 m2/g, and very fine sand 0.1 m2/g. Once you get into the clays (diameter < 0.002 mm) the SSAs are huge; montmorillinite is about 800 m2/g. So an Alka Seltzer of mass 3.278 g and ground "fine" has a total surface area of 0.200 m2 (which is a whopping 200000 mm2). Whew, that's big!
Lastly, as the tablet dissolves and the chemicals react, its surface area decreases so that factor is no longer controlled. As well, as it dissolves it breaks up into small pieces so you have another problem, and it is hard to see when it is all dissolved as there are bits of gunk floating around. Lastly, sometimes the tablet floats on top of the bubbles on the surface of the water and the tablet can't get to the water.
Reaction Rate and Surface Area - II
Schools often use soluble aspirin tablets as they are smaller and cheaper. Here are some results to get you thinking. These are the averages of 16 groups from my Year 8 class; mass of tablet 1.240 g, diameter 17.16 mm, thickness 3.70 mm, volume of water 50.0 mL, beaker 100 mL, gentle stirring. The graph to the right is drawn from research data on the reaction rate of powdered limestone (calcium carbonate) as a function of particle size (and hence surface area). The numbers on the lines show each of the four particle sizes used (in µm).
Reaction Rate and Surface Area - III
If you want to get away from fizzy tablets here are a few other surface area experiments you could try:
Reaction of marble and acid You may have also tried this using marble chips and calcium carbonate powder. But how to measure the surface area? My suggestion is to get some marble tiles from a tile shop and cut them into strips with a masonry blade on an angle grinder. If you have five strips you can break one in half, one in quarters and so on. Using a Vernier calliper it will be easy to measure surface area. The method id quite straightforward after that. Nevertheless, give some thought to how you will control temperature, and how much acid you will need, and what concentration so that it is not the limiting reagent. Lastly, what will you measure for reaction rate: time taken for the chips to dissolve, or a flask on a balance with recordings taken every minute...and so on. A great technique for an EEI would be to pour off the acid after a given time and titrate it with standardized NaOH solution to see how much acid (and hence marble) was used.
Zinc and acid This is a lot simpler as you can cut zinc sheet with scissors. You'll still need Vernier callipers to measure the dimensions, so the error will be larger than with the marble but it may be a lot simpler to make sense of. Hmmm, what concentration acid will I need? Will it heat up? Displacement Reaction This has the potential to be a great (not just a good) EEI. More reactive metals will displace less reactive ones from solution. If you've done the Redox unit in Chemistry you will be aware that a reactive metal like zinc, when placed in an aqueous solution of a salt of a less reactive metal (eg Cu as CuSO4 solution) a reaction will occur. The zinc will dissolve to form Zn2+(aq) ions, and the Cu2+(aq) ions in the CuSO4 solution will accept electrons from the zinc to become copper metal. The solution starts off as a bright blue colour due to the presence of Cu2+(aq) ions but as these are consumed the solution gets less and less blue. If you have access to a spectrometer then this would be easy to measure. You could make up a solution of known concentration (recall that CuSO4 is actually in the pentahydrate form CuSO4•5H2O when working out molar masses). Just measure the absorbance of it (the lambda max for the copper solution is 740 nm) on the spectrophotometer and from a graph (assuming the Beer-Lambert Law is still working from 1852) and if you know the absorbance you can work out the concentration of the copper ions. Your teacher may suggest that you prepare a range of standard solutions of Cu2+ to produce your own calibration curve. Now, using a zinc strip and ones cut into halves, quarters and so on (all measured with Vernier callipers) you can place them in identical copper sulfate solutions and measure the change in blueness after say an hour or a day. What a great EEI. I wish I was young again - I'd do it.
Zinc and acid This is a lot simpler as you can cut zinc sheet with scissors. You'll still need Vernier callipers to measure the dimensions, so the error will be larger than with the marble but it may be a lot simpler to make sense of. Hmmm, what concentration acid will I need? Will it heat up? Displacement Reaction This has the potential to be a great (not just a good) EEI. More reactive metals will displace less reactive ones from solution. If you've done the Redox unit in Chemistry you will be aware that a reactive metal like zinc, when placed in an aqueous solution of a salt of a less reactive metal (eg Cu as CuSO4 solution) a reaction will occur. The zinc will dissolve to form Zn2+(aq) ions, and the Cu2+(aq) ions in the CuSO4 solution will accept electrons from the zinc to become copper metal. The solution starts off as a bright blue colour due to the presence of Cu2+(aq) ions but as these are consumed the solution gets less and less blue. If you have access to a spectrometer then this would be easy to measure.
You could make up a solution of known concentration (recall that CuSO4 is actually in the pentahydrate form CuSO4•5H2O when working out molar masses). Just measure the absorbance of it (the lambda max for the copper solution is 740 nm) on the spectrophotometer and from a graph (assuming the Beer-Lambert Law is still working from 1852) and if you know the absorbance you can work out the concentration of the copper ions. Your teacher may suggest that you prepare a range of standard solutions of Cu2+ to produce your own calibration curve.
Now, using a zinc strip and ones cut into halves, quarters and so on (all measured with Vernier callipers) you can place them in identical copper sulfate solutions and measure the change in blueness after say an hour or a day. What a great EEI. I wish I was young again - I'd do it.
Corrosion of Copper by Sulfuric Acid It may seem surprising but there are almost no journal articles by chemistry researchers on the effect of surface area on reaction rate - in industry or academia. Those that do relate to the area of catalysts rather than the main reactants (but that does suggest another EEI topic). The most recent paper as a stimulus for a high school chemistry EEI is one by industrial chemists Glenn Damon and Ray Cross from the Michigan College of Mining and Technology, Houghton, Michigan published in Industrial and Engineering Chemistry journal V28 (2) in February 1936. They reacted sulfuric acid with small squares of copper placed 2 cm under the liquid surface. However, to manipulate the surface area variable they varied the surface area of the solution exposed to the atmosphere. You could prepare a small circular piece of polystyrene foam (with a hole cut in the middle) and float it on the surface of the acid. This will give limited access of oxygen to the solution and hence limit the corrosion of the copper. It is a neat experiment and may give you a few ideas. Click here to download it.
Smartphone colorimeter
This is not an EEI suggestion but a way of measuring solution concentrations colorimetrically. It could be used for any solution whose colour intensity is a measure of its concentration, eg blue copper ions. It may be useful if you don't have access to a colorimeter (as above), or even a cheaper one that can be used with a data logger (such as Vernier connected to a LabQuest as shown below) then a smartphone app may be the answer.
To test out the smartphone app, I made up some standard copper ion solutions of 4.0 g of copper per 100 mL (0.6 M) using copper (II) nitrate (Cu(NO3)2.3H2O). I then diluted this to make a series of dilutions from about 0.24 M up). The Color Grabber app on an Android, or Color Picker app for the iPhone work well and reports HSV values. The H is for Hue and reports it on a scale of 0-360° which can be used as an index of absorbance. The "S" is for Saturation in %
Solubility of salts in water and alcohol
A study of salt solubility in different solvents is very important for many industrial applications. More particularly, a knowledge of accurate solubilities is needed for the design of separation processes such as extractive crystallization or for the safe operation of different processing units such as distillation columns, absorption units, and extraction plants.
Aqueous electrolyte solubility is generally available for many salts, but aqueous-organic mixed solvents data is very scarce, obsolete, or not available at all. An EEI based around this sounds like a good idea. You could try salts NaCl, KCl, and NaBr and solvents methanol and ethanol. To avoid water salt contamination, salts could be dried at 100°C in a drying stove for at least 2 days before use.
All you need do is to prepare a saturated solution at a desired temperature (with undissolved solid on the bottom) and keep it stirred for an hour or two. Let settle and take a sample of the supernatant liquid, weigh it, and evaporate the water (on a hotplate to dryness and then in an oven at 120°C for a day until mass is constant). Might take a day or two.
Then you could try binary mixtures of solvents. Have a look at these results*:
*Click here for a good article: "Solubility of NaCl, NaBr, and KCl in Water, Methanol, Ethanol, and Their Mixed Solvents" in the Journal of Chemical and Engineering Data, Vol. 50, No. 1, 2005, p29-32. The authors are Simão P. Pinho and Eugénia A. Macedo, Laboratory of Separation and Reaction Engineering, Departamento de Engenharia Química, Faculdade de Engenharia, Rua do Dr. Roberto Frias, 4200-465 Porto, Portugal. Thanks guys.
Cetyl alcohol and water evaporation losses Billion of litres of water normally lost each year through evaporation from the nation's waterways - including reservoirs, lakes and dams. Evaporation from Australian water bodies ranged from 1.3 m to 1.9 m per year (Brisbane is 1.6 m per year), with an average evaporation rate of 0.5 litres per day per square metre, or 5000 litres per day for every one hectare of open water. For a water body covering about 100 hectares of open water - a medium sized reservoir or dam - approx 190 million litres of water (or 75 Olympic-sized swimming pools) is lost every year through evaporation - this is equivalent to the annual consumption of 380 typical Australian households.
However, recent trials in Australian conditions by several council/municipal water managers and commercial cotton farms confirmed evaporation savings of about 30% using various long-chain alcohols. These alcohols such as cetyl alcohol - also known as hexadecanol, CH3(CH2)15OH - develop an invisible film (or monolayer) on the water surface, creating a barrier that limits the escape of water vapour. Chem-Supply in Australia have cetyl alcohol (Code: CL044-500G) for about A$39 per 500 gram bottle Lab Reagent (LR) grade (plus $27.50 for 3-5 day delivery) but a commercial grade is also available elsewhere (but in big quantities).
Sometimes it may take a while to get so cetyl-stearyl alcohol would be fine (you can often get this from places that sell home-made soap suplies). It is merely a mixture of cetyl alcohol (C16) and stearyl alcohol (C18). This suggests a good EEI. You could look at the evaporation of water from an open container, with and without a monolayer of long-chain alcohol. It obviously is not water soluble so you would need to make a solution by using a different solvent (try ethanol). The independent variable could be the amount of alcohol (per sq metre) or the thickness of the film, and the dependent variable could be the amount of water evaporated. To get reasonable evaporation rates you really need to use an electric fan blowing across the top of the water (for at least 24 hours).
How you measure the change in water level is up to you (by mass, by height). You'd get even better results if the ambient temperature was warm (eg in a fume cupboard with the heating lamps on). Research being done by Ian Craig, Erik Schmidt and Michael Scobie from the National Centre for Engineering in Agriculture (NCEA), University of Southern Queensland (USQ) into the use of these monolayers can be downloaded here. I based the idea above on an article "Alternative methods for the reduction of evaporation: practical exercises for the science classroom" by Peter Schouten and colleagues from Griffith University's School of Engineering, Gold Coast, Australia. Peter has allowed me to make it available for download here. It was published in Physics Education (2012, V47, No 2, p 202-210)
Heating up gases You would have seen how gases expand when they are heated. Your teacher may have heated a flask with a balloon on the top to show it expanding; you may have seen a balloon shrink when dipped in liquid nitrogen at -198°C; and it is the principle behind how hot air balloons work. In class you would have called the law describing the relationship between temperature and volume Charles's Law or perhaps Amonton's Law (V ∝ T when T is in kelvin and P and n are kept constant). There could be a great EEI in revisiting this relationship. There is no point in just verifying it as this has been done a million times. What you want to do is to extend the investigation of this law to look at the impact of changing variables and to consider allowing for errors.
The diagram below shows a setup that may be useful. It really just show the connection of two things: a flask with a sidearm (maybe a Büchner flask) and a graduated glass syringe. The exact positioning is something you should determine. Glass syringes are precision-made with low friction between the plunger and the barrel (unlike plastic ones that have high friction). Your should have some in the chem lab and if not they are reasonably cheap (about $50 for a 100 mL one). You need to introduce a gas (eg CO2) into the flask and surround the flask with water in a beaker on a hotplate. As it slowly heats (I mean slowly, maybe 20°C to 80°C over 40 minutes) the gas expands and the syringe is pushed out. With the syringe on it's side there is no need to worry about the weight of the plunger. You could compare gases - oxygen, nitrogen, hydrogen for example.
But how to get samples of these gases? You may have cylinders but you could produce H2 and CO2 by reaction (or let some dry ice sublimate); let some liquid nitrogen evaporate (or remove oxygen from air). And why not propane (BBQ gas) or butane (cigarette lighter fluid)? Remember that balloon gas is not just helium - it has 3% air mixed in with it. The main point is that the law holds for ideal gases but at atmospheric pressure and room temperature they won't be that ideal. And is the deviation from ideality dependent on the molar mass of the gas, or whether it is polar or non-polar, and where on earth do you get a polar gas from (HCl is too dangerous)? What range of temperatures will you use (consider liquid nitrogen, dry ice). What value will they give you for absolute zero when the V/T graph is extrapolated? How do you draw the line of best fit (is least-squares the best, does it give you the most accurate value for absolute zero?). And what is the volume of the gas in the apparatus? And what is the best way to measure temperature (of the gas as in the diagram, or of the water surrounding it)?
Perhaps the temperature of the gas in the flask is the water temperature and the temperature of the gas in the syringe that of the surrounding air (work out a weighted average). And how do you control atmospheric pressure (do you have a barometer, or perhaps get the data from the meteorological bureau website). What a fabulous EEI. I must put this on the Physics EEI webpage as well.
Aging (fermenting) orange juice Here's a comment off a health food blog from a guy called Vincent: "I was too lazy to wash out a 2 L carton of Tropicana orange juice after dinner last night. I go to wash it out today and the carton was bulging quite noticeably. Those crazy orange juice fermenting bacteria work fast! The carton let out a nice puff of air when I opened it up and it tastes so sour." What has happened here? Orange juice has a lot of natural sugars in it. Bacteria love it if you let them get in. The refrigerator only slows growth of bacteria, it doesn't kill them.
These bacteria aren't necessarily the kind that make you sick, but they will start to grow and will begin to break down the orange juice. It will start to ferment-if you taste it it will be bubbly and will taste sour from the build up of acids - possibly acetic acid from the alcohol. Is there an EEI in this? There certainly is and it needs careful consideration about controlling variables and you need to think about what acids are present besides citric and ascorbic. What variable might you manipulate? The total acidity can be measured by titration with sodium hydroxide.
Cheesemaking Here's one from Gary Turner at St Mary's Catholic College South Burnett. Most major newspapers have a life-style section in which appear columns about cheeses and wines. Australia has several small cheese-making plants in which hand-craft is as important as technology. Cheese-making is a promising industry within the local region. A closely related product, amenable to student-investigation is the making of sour-cream, which is commonly used in several fast-foods of interest to teenagers.
First: You will be following a 'standard' procedure for making a simple cheese (e.g. ricotta) or sour cream to give you the background skills and chemistry involved in making a cheese, and to explore the factors involved. (This can be done as a group).
Second: You are then to select another cheese that interests you and individually make this cheese and explore the factors that affect the result (e.g taste and texture and hardness). This section of the work will also require you to define which factors you can reasonably test in a school-laboratory, and which variables in the production that you can vary.
Third, you are to compare your cheese to a similar commercially available cheese and report on the differences and likely causes of that difference. (The factors that you can compare will be those that you have defined in the second section above). A copy of this cheese EEI is available for download here. A useful video from the ABC TV Landline program maybe worth watching.
It shows Yr 12 science students from Sandgate Sate High School (Queensland) making cheese under the guidance of master cheesemaker Russell Smith and Chemistry teacher Alison Turner. The link to the video is here. Another Landline report shows the students entering their cheeses into the Royal National Association show ("The Ekka"). See "Lateline Masterclass" here. Remember - making cheese does not make an EEI.
Lactic acid and the fermentation of milk Lactic acid forms in milk due to the action of fungi and bacteria acting on the lactose sugar. The most important lactic acid producing bacteria is Lactobacillus. The presence of lactic acid, produced during the lactic acid fermentation is responsible for the sour taste and for the improved microbiological stability and safety of the food. A good EEI might be to investigate the factors influencing the rate of formation of lactic acid upon the addition of some starter bacteria (eg plain yoghurt). I won't say what they are but a couple of the following are suspects: heat, amount of bacteria added, light, access to air, shape of container, sugar concentration, initial pH, amount of fat (normal, low fat, skim), degree of agitation, and so on. Start with heat.
The acidity in milk is sometimes measured by titration with a 0.1 M NaOH solution, and indicates the consumption of NaOH necessary to shift the pH-value from 6.6 (corresponding to fresh milk) to a pH-value of 8.2 - 8.4 (phenolphthalein end point). People sometimes wrongly assume that the titratable acidity is due to lactic acid - an organic acid with the formula CH3-CHOH-COOH. However, fresh milk contains practically no lactic acid and the consumption of NaOH is used to change the pH-value of the following components: carbon dioxide, citrates, casein, albumin and phosphates which gives the appearance of a lactic acid concentration of about 0.13% The determination of "acidity" in fresh milk by means of titration is therefore more a measure of the buffer action of milk than anything else. If you try to calculate the theoretical pH of milk based on the titratable acidity (using the Ka for lactic acid), you will get stupid results - like a pH of 2.5 for milk.
In an EEI, it is likely that you want to talk about the "developed acidity", which is the result of bacterial activity producing lactic acid during milk collection, transportation, and processing. In order to avoid the uncertainties about the degree of titratable acidity or developed acidity, it is necessary to use a different method for determining lactic acid. A rapid colorimetric method for the quantitative estimation of lactic acid in milk is available but way beyond the facilities of a high-school lab. The only way out of this conundrum is to measure "titratable acidity" (rather than calling it "lactic acid concentration") but acknowledge the errors and subtract the initial "acidity" from the subsequent values obtained during the experiment. Be careful if you intend to measure titratable acidity as a function of time eg "time elapsed" (rather than just as a function of some manipulated variable (such as temperature). See the note that follows. As a rough guide, one of my students measured the titratable acidity of milk as 0.0288M as lactic acid (7.10 mL titre) at the start, and on day 7 the value was 0.0815M (15.65 mL titre). The in-between values did not give a linear graph; it was much more exciting than that.
Note about identifying variables: "time elapsed" can be a controlled variable or independent variable (or both) in this experiment (and others that involve collecting data over a period of time). CASE 1: In the fermentation experiment you may, for example, choose to have the "temperature" as the independent (manipulated) variable (say 0°C,10°C, 20°C, 30°C...) and "titratable acidity" as the dependent variable. If these are measured just once, say after 1 week, then "time" is a controlled variable (along with initial pH, sunlight, sugar concentration, aeration, exposed surface area etc). You could prepare a graph where you plot "titratable acidity" (y-axis) and temperature (x-axis) and there will be one line.
CASE 2: However, "time" can be an independent variable as well. You use the "temperature" as the independent variable but if you measure the dependent variable (titratable acidity) every week at 0, 1, 2 and 3 weeks then you really have two experiments in one. There are two independent variables: "time" and "temperature" but they can be examined separately. A plot of titratable acidity (y-axis) vs time (x-axis) would show 4 lines (if you used 4 different temperatures). This would be most valuable as it would show you the fermentation rate at each temperature.
You could prepare another graph where you plot titratable acidity (y-axis) and temperature (x-axis) to get 4 lines (one for each weekly measurement including the titratable acidity at t=0). This would be harder for you to visualise and interpret however. The two graphs together could be analysed "... to identify relationships between patterns, trends..." IP3 (VHA) and "analysis and evaluation of complex scientific interrelationships" (EC1, VHA). The two graphs provide stronger evidence for inter-relationships than either graph alone.
There is an increase in the concentration of H+(aq) ions during this discharge and this can be monitored by titration with a base such as sodium hydroxide. To discharge the battery rapidly but steadily the students Olivia and Kayla at Moreton Bay College used 12V car lightbulbs across the terminals. They asked - is the change of [H+] proportional to the duration of discharge? Perhaps the rate of discharge as well as the duration important. Should they monitor voltage and current as well? You decide. My thanks to their teacher Mrs Cathy King for welcoming me into her lab.
Thermal stability of sodium bicarbonate (why my cake didn't rise) Sodium hydrogen carbonate NaHCO3 also known as sodium bicarbonate or "bicarb of soda" - is an important component in various pharmaceutical drugs (tablets, capsules, syrups) and cooking (scones, cakes). Because of its widespread use, the stability of sodium bicarbonate in solid state, both as a raw material and as a formulation component, is of high interest to the pharmaceutical and food technology scientists.
When sodium bicarbonate is stored as a powder, it degrades over time to carbon dioxide and sodium carbonate after absorption of moisture at lower temperature, or degrades directly to carbon dioxide and sodium carbonate without absorption of moisture at elevated temperature (Shefter et al., 1975 - see below). Therefore, it is critical to maintain appropriate temperature and relative humidity during the storage of the raw material and finished product as well as during manufacturing. Sometimes you want it to breakdown - but only when you're ready: cooks rely on the breakdown of "bicarb" at high temperatures into carbon dioxide to make cakes and scones rise.
A good EEI would be to examine the conditions for the thermal breakdown of NaHCO3. You could do this several ways: one would be to take some samples of NaHCO3 and hold them at various temperatures from room temperature to the maximum temperature of your oven and titrate a solution of the sample against HCl after a fixed time (eg 1 hour). Because we have two substances in the solid mixture (NaHCO3 and Na2CO3) there will be two analytes in the solution of the mixture, namely HCO3- and CO32-. The problem is: how do you tell how much of each there is. Here are the reactions so you can see what goes on in neutralisation: Carbonate titration: The Na2CO3 present in solution will react with the acid as the titration proceeds until it is all converted to NaHCO3 (aq): Na2CO3(aq) + HCl (aq) → NaHCO3 (aq) + NaCl (aq) Bicarbonate titration: As you add more acid, the bicarbonate that was present initially and the bicarbonate produced by the first titration will react: NaHCO3(aq) + HCl (aq) → NaCl (aq) + CO2(g) + H2O (l)
What we need is two different indicators, one to indicate the endpoint for the reaction between H+ and CO32- and the other to indicate the endpoint for the reaction between H+ and HCO3-. The first is phenolphthalein and the second is bromocresol green. To calculate the amount of bicarbonate in the mixture, you subtract the amount of carbonate from the total amount of bicarbonate. The titration is quite complex because of the dissolved CO2 generated which you have to boil off. See Oliver Seeley's How to Titrate Carbonates webpage for some great hints and cautions.
ALTERNATIVES: 1. Alternatively you could titrate samples taken over a range of times (10, 20, 30…etc minutes). You'd have different graphs for the two approaches but what a great EEI. Is commercially available sodium bicarbonate different to the analytical reagent from the chem lab? Be careful if you buy "baking powder" rather than "baking soda"; baking powder has starch and sodium acid pyrophosphate added to give more gas.
I have attached two scientific papers that may provide some ideas: one is Effect of relative humidity and temperature on moisture sorption and stability of sodium bicarbonate powder by Kuu, Chilamkurti & Chen (1998) from the International Journal of Pharmaceutics (1998), and another A Kinetic Study of Sodium Bicarbonate by Schefter from the journal Drug Development Communications (1975). They are heavy going but this is Year 12 Chemistry after all.
Breakdown of Antacid Tablets
Instead of investigating the thermal breakdown of sodium bicarbonate in baking soda (as above), you could consider the effect of heat on antacid tablets. These tablets are designed to neutralise stomach acidity so they are basic salts, usually calcium carbonate (eg Quick-eze or TUMS). The process and analysis would be the same as above. You could try Mylanta tablets (magnesium hydroxide and aluminium hydroxide) but Gaviscon may be tricky (aluminium/magnesium trisilicate). Remember, in an EEI you are not just trying to simulate real life; in this case you are trying to extend the understanding of carbonate breakdown to a bigger range of temperatures than you would find at home or in a car glove-box. That's why you would use the lab oven to get high temperatures for the trials.
Soft Drinks and Tooth Decay
Soft drinks consumption has risen dramatically over the past 40 years and so has the resulting incidence of rotting teeth and osteoporosis. Does this sound like a fun context for a chemistry experiment? The photo to the left shows perfect teeth. If I used a photo of rotting teeth you would feel sick.
A soft drink such as lemonade or Coca-Cola is a drink that does not contain alcohol, as opposed to a hard drink, which does. Australians consume about 300 mL of soft drink per day on average but amongst 14-16 year olds the figures are 1000 mL for males and 500mL for females. Soft drinks are about 10% sugar so a young male typically consumes 27 teaspoons of sugar per day in soft drink; a girl, about half that. Just one can of soft drink has about 10 teaspoons of sugar in it. The resultant obesity (fat) epidemic is attributed in part to soft drinks. Health risks from over consumption include diabetes, kidney stones, obesity, osteoporosis, and tooth destruction.
Tooth decay is partly from the bacteria feeding on the sugar but also from the acids reacting with the tooth enamel. The citric or phosphoric acid in soft drink dissolves the calcium out of the enamel leaving a softened matrix for bacteria to enter the teeth and cause wholesale carious (tooth) destruction. So drinking sugar-free (diet) soft drink is not the answer.
A good EEI may be to look at the effect of drink acids on teeth. Teeth are a form of hydroxyapatite Ca5(PO4)3(OH) but you can simulate this in the lab with calcium carbonate (marble chips). The problem is: you need to control the type of acid, whether it is phosphoric acid as found in cola drinks, or citric acid as found in lemonade. A study by Fraunhofer and Rodgers (2004) found that the rate of enamel dissolution of teeth was not dependent on pH but may be affected by titratable acidity. Remember that weak acids (phosphoric, citric) are not fully dissociated in water (so their pH is not that low) but they gradually release more hydrogen ions as they react. The "titratable acidity" will be a measure of this. Citric acid, for instance, is a tribasic acid which releases its H+ ions in four steps. It has a reversible reaction with CaCO3 and the reaction is controlled by diffusion of reaction products away from the 'tooth' surface; thus, consider keeping it stirred.
As a trial, I took a 1 cm3 cube of marble and placed it in 200 mL of 7.5%w/v citric acid solution (pH 1.8) at 50°C (with stirring) and after 60 minutes it had lost 0.30 g. Why not make up a synthetic soft drink from phosphoric or citric acid. What concentration will you choose? How does the reaction rate or the extent of the reaction vary with concentration? Does temperature have much effect on the rate? Does the product - calcium citrate or calcium phosphate - impede the progress of the reaction; that is, how soluble are the products (one is 4 times as soluble as the other). How do you measure the progress of the reaction (amount of carbonate consumed or change in titratable acidity of the solution)? Oh, the possibilities are endless. And you can drink the left-over Coke and rot your teeth a bit more at the same time. A perfect EEI.
CCA Treated Timber Leaching The most common wood preservative in Australia is chromated copper arsenate, or CCA which produces a greenish colour in the wood (see photos below). Concerns sometimes arise over the use of treated lumber in vegetable beds. In the USA, CCA preservative was phased out in 2003, for virtually all residential uses, including fencing, decking, children's play equipment and raised garden beds. Two other products, ACZA (ammoniacal copper zinc arsenate) and ACQ (ammoniacal copper quat) have replaced CCA and in the USA may be used for raised bed construction. We are told that CCA, ACZA, and ACQ may be safely used to construct vegetable beds. But who would believe that?
The Queensland Environmental Protection Authority ensures that when CCA fences or posts are burnt by bushfires in national parks the ash is buried as a contaminant to keep away from the public. If you have access to a spectrophotometer, you could investigate the amount of leaching from CCA pine by taking some CCA pine shavings (caution - gloves) and immersing them in water. You could vary conditions such as pH, amount of stirring, time in contact. As usual, a risk assessment will need to be submitted before you start.
Anodising Titanium Titanium is an amazing metal. It is strong, light and corrosion resistant. It can be alloyed with many metals to increase its range of applications for industrial, aerospace, recreational, and emerging markets. Its behaviour when anodised is remarkable. Anodizing titanium produces an oxide coating which generates an array of different colours, making it appealing for art, costume and body piercing jewellery and architecture (eg Guggenheim Museum). The color is an interference effect much like that in a soap bubble. The anodised colour depends on the voltage (see chart below). You could investigate the relationship between colour and voltage using different electrolytes. The big problem is getting 100 volts. Connecting a heap of 9v batteries in series might do the trick.
Iron filings in fortified cereal A healthy adult needs about 18 mg of iron each day. Dietary iron is found in large amounts in organ meats such as liver, kidney, and heart. It is also present naturally in egg yolks, some vegetables, and shellfish. In these foods, iron is typically present as Fe (III) ions. Our body absorbs iron in the small intestine in the form of Fe (III), which then is reduced to Fe(II). Under normal conditions, our body absorbs only 5-15% of the iron in the food that we eat. Cereals are fortified with food grade iron filings as a food supplement. This iron is metallic iron (Fe).
In the stomach the metallic iron is oxidized and eventually absorbed through the small intestine. You can see the iron if you pass a magnet over a slurry of breakfast cereal. I used Sanitarium's Light 'n' Tasty but any will do (see my centre photo below using a 100mm macro lens). The question is - how to make an EEI out of this? You need to do more than measure and compare the amount of iron in breakfast cereals. A good EEI would be to investigate methods of extraction of the iron perhaps involving the use of a magnetic stirring bar before analysis. Could you dissolve the metal in acid? Is all the iron in the form of elemental iron (filings) or is there some natural iron compound present?
Polyurethane foam Polyurethane is a synthetic polymer widely used in flexible foam seating, seals and gaskets, tyres, bearing bushes, adhesives and sealants. The type you may be familiar with from school (if you made a polyurethane foam mushroom) is called a 'rigid foam' and is used for insulation panels and surfboards. They are made from two monomers - isocyanate and polyol. In 1984 water was accidentally introduced into a reaction mix and the first foam was made.
A good EEI could be to look at the conditions required to produce the different densities of rigid foam. You could use equal amounts of the monomers and try them with different temperatures or different amounts of stirring. You could even try adding more water to the polyol monomer. You could even try making a variable density foam by placing the reaction vessel (plastic cup) on a cold surface. The main thing is to explain why you'd want a particular density and hypothesise how it could be achieved. It will be heaps of fun.
Alcohol-water mixture: concentrations and the contraction of volume When you mix ethanol and water together the final volume is less than the sum of the separate volumes you started with. This shrinkage is known as 'volume contraction' and is due to the strength of the hydrogen bond. Such a bond is strong in water but weaker in alcohols, however, when a mixture is made the dipole-dipole forces tend to make the alcohol-water clusters small. Technically, we could say "departures from Raoult's law are often found in liquid mixtures resulting in volume nonadditivity". In practice, this contraction can have vital consequences.
Medical researcher know that alcohol absorption into the bloodstream and the resultant volume contraction can upset the plasma concentration of various biochemicals and lead to all sorts of complications. A good EEI would be to measure the volume contraction of various mixtures of ethanol and water 25:5, 50:50, 75:25 and so on to see how the percentage contraction varies (the point of maximum contraction could be found). And if it is true that the effect is due to H-bonding, should the contraction be different for alcohols exhibiting weaker or stronger dipole-dipole forces (eg the monohydroxy alcohols: methanol, 1- and 2-propanol, tert-butanol)?
I wish I was doing an EEI - this one would be great. I'd be looking at the work of our famous friend Dmitri Mendeleev (of Periodic Table fame) who found that a 1:3 mixture gives the biggest contraction (see Analytical and Bioanalytical Chemistry, 2009, V 395 (1) 2009, p7-8).
Alcohol-water mixture: temperature and the contraction of volume In the suggestion above, the investigation of volume contraction of ethanol water mixtures was suggested. Of equal interest would be the effect of heat which is known to affect the strength of the H-bond; so you could see how stable the % contraction was over a range of temperatures. Safety warning: alcohol water mixtures can burn even when the amount of alcohol is less than 50% - and especially at higher temperatures.
As well, if the contraction effect is due to H-bonding, shouldn't the contraction be different for alcohols exhibiting weaker or stronger dipole-dipole forces (eg methanol, propan-1-ol, propan-2-ol and methyl propan-2-ol)? As a matter of interest, mix together CS2 and ethyl acetate and you get volume expansion (but CS2 is too dangerous for high school experiments).
Capillary action Capillary action is the tendency of a liquid to rise in narrow tubes or to be drawn into small openings such as those between grains of a rock. Capillary action, also known as capillarity, is a result of the intermolecular attraction within the liquid and solid materials. A familiar example of capillary action is the tendency of a dry paper towel to absorb a liquid by drawing it into the narrow openings between the fibers. Some liquids exhibit more capillarity than others; for example, there is a big difference between water, salt water, ethanol and hexane. A good EEI would be to compare capillary action (between two microscope slides; see below) for polar and non-polar liquids, or non-polar ones of different density, or salty water vs distilled water, or as a function of temperature or capillary gap. You could also somehow use capillary tubes (see below). The possibilities are huge, but don't get too carried away.
Browning of apples Apples turn brown when peeled and exposed to air. This discolouration is due to a process called enzymatic oxidation and is catalysed by the enzymes present in the apples. The enzyme polyphenol oxidase (phenolase), in contact with oxygen, catalyzes one step of the biochemical conversion of plant phenolic compounds to brown pigments known as melanins (brown, like a suntan). It occurs at warm temperatures when the pH of the plant material is between 5.0 and 7.0. Browning can be stopped!
Vitamin C, being a highly reactive anti-oxidant reacts with the O2 in the air, preventing/slowing down the enzymatic oxidation of the apples. Another way to reduce browning is to lower the pH in order to inactivate the enzyme. Ascorbic acid is used commercially to prevent enzymatic browning as it acts as both an acidulant and antioxidant. To make an EEI out of this you could test the browning when controlled volumes of acids of various [H+] are used, and then with ascorbic acid of known [H+] to see how much is due to the antioxidant property. Temperature could also be assessed. It is also said that Fe and Cu speed it up but Ca2+ slows it down. Question: how will you measure the browning? Remember this is chemistry not MasterChef. Laboratories use a reflectance spectrophotometer at a wavength of 400 nm to measure the degree of browning but it is unlikely you'd have access to one of those. If you have a regular (transmission) spectrophotometer you could take a sample of your browned apple, blend it up, filter it and measure absorption at 400nm. You could compare various treatments (providing the surface area of the same was controlled). Failing this, perhaps let an apple go brown and take a photo and make that your official "brown" (or make a range of photos on a scale of 1-10 from 'not brown' through to 'very brown".
Effect of catalyst concentration on reaction rate: enzymes You will have read that catalysts are substances that speed up reactions but that they are only needed in small quantities. A great EEI would be to test this proposition and to see if there is a quantitative relationship between amount of catalyst and reaction rate. Maybe once you have added sufficient catalyst then adding extra makes no difference. A good catalyst for this experiment would be a biological catalyst - an enzyme.
Enzymes, like other catalysts, catalyze reactions by lowering the activation energy necessary for a reaction to occur. The molecule that an enzyme acts on is called the substrate. The enzyme molecule is unchanged after the reaction, and it can continue to catalyze the same type of reaction over and over. The enzyme catalyze will speed up the breakdown of hydrogen peroxide (the substrate) into water and oxygen. Say you took 10 mL of 3% peroxide solution, added some water, and added different amounts of catalase to each, you may get different reaction rates. The catalase can be made up into a suspension with water and different amounts added dropwise (0, 5, 10, 20 etc) to the peroxide solution. What to measure? The simplest way to start making measurements is to stop the reaction by adding sulfuric acid as this destroys the enzyme's functioning and the reaction stops.
As this is a chemistry EEI should look for standard chemical methods of analysis. No doubt you've learnt about titrations, so to see how much peroxide was used up you could titrate say 10 mL aliquots of the solution against a standard KMnO4 solution in the burette. Another method may be to use a pressure sensor in the neck of the flask and hook it up to a datalogger. If you want to examine another variable, you could hold the amount of catalase constant and vary the temperature, or vary the pH (I said before that adding sulfuric acid would destroy the enzyme, but how sensitive is it to pH).
Effect of catalyst concentration on reaction rate: copper catalyst This is a tricky one but there is so much to explore. It is a bit more complicated because it involves reaction rates and redox theory - but the method should be quite simple. When you add hydrochloric acid to zinc it reacts at a certain rate; however, if a piece of copper touches the zinc then the zinc reacts much faster. Why? And does this means copper is a "catalyst" because it speeds up the reaction (doesn't it have to appear unchanged at the end - well is it)?
An electrolytic cell is created where the more reactive metal (Zn) is the anode and Cu is the cathode, with the HCl acting as an electrolyte, so hydrogen ions are likely to be reduced to hydrogen gas at the copper electrode. Without the copper, there is no obvious electrolytic cell, so hydrogen gas would have to be produced by the direct reaction between zinc atoms and hydrogen ions at the surface of the metal and this is slower. A great EEI could be investigating the effect of varying the amount of copper in contact with the zinc.
A great way to get copper in close contact with the zinc is to deposit copper metal directly on to the zinc by a displacement reaction. To do this you would dip the piece of zinc into a solution of copper sulfate and a coating is immediately deposited. If you dipped identical pieces of zinc strip into a copper sulfate soution - but each to a different depth (0 cm, 1 cm, 2 cm ....) and for the same time you would get different areas of coating. Then react the strips individually with hydrochloric acid and measure the rate by whatever method you like (change in mass due to lost hydrogen, amount of gas produced, temperature change, titration the final solution against NaOH).
Deactivation of pineapple enzymes If you've ever tried to make a jelly with pineapple or kiwifruit in it you may have been sorely disappointed. It may not set because the enzyme catalyst has played up. All living cells produce enzymes which catalyze metabolic reactions. The enzyme that you could investigate in an EEI is one that is produced in pineapple and hydrolyzes certain kinds of proteins called gelatins. Gelatin used in jelly is derived from skin, bones, and/or connective tissue of animals (vegetarians have to use agar type jellies). Gelatin proteins, when dissolved in hot water and allowed to cool, form a semi-solid or gel state; hence the name gelatin (or gelatine). Hydrolyze, here, refers to breaking up the protein polymer in such a way as to prevent its forming this gel state. The hydrolyzing enzyme from pineapple is denatured (destroyed) by heat; but not freezing - I don't think.
Enzymes can also be denatured by changes in pH, detergents or radiation. You could take some pineapple and subject it to different heat treatments and see its effects on the gelatine. You have to make up a device and method for testing gelatine that allows replicable and meaningful testing. I recall using the Bloom Strength test at Golden Circle - it was the mass in grams required to press a 12.5 mm diameter plunger 4 mm into the gel. If you did it at home you could even eat the results.
pH of vinegar solutions If you've completed an acids and bases unit you will be aware that strong acids and bases, like HCl and NaOH respectively, dissociate fully. However, weak acids and bases only partly dissociate and the equilibrium constant (Ka or Kb) gives a measure of this dissociation. The quantitative behaviour of acids and bases in solution can only be understood if their Ka (or pKa) values are known. Such knowledge finds applications in many different areas of chemistry, biology, medicine, and geology. For example, many compounds used for medication are weak acids or bases, and a knowledge of the pKa values can be used for estimating the extent to which a compound enters the blood stream.
Acid dissociation constants are also essential in aquatic chemistry and chemical oceanography, where the acidity of water plays a fundamental role. In living organisms, acid-base homeostasis and enzyme kinetics are dependent on the pKa values of the many acids and bases present in the cell and in the body. Here's a suggestion for an EEI: if you make up a solution of known concentration of say acetic acid CH3COOH, and then measure it's pH, you can calculate the Ka using standard formulas. No doubt there will be an error but your EEI could be to investigate the source of this error and try ways to minimize it (is it the formula, the calibration of the pH meter, the dilutions, the temperature?).
You could see if the error varies with starting concentration of the acid [HA] and also look at the effects of temperature. A comparison of several weak acids may also be revealing. How accurate is the pH meter for dilutions of strong acids? And then there are the bases. The possibilities are endless.
Water retention in disposable nappies Today's state-of-the-art disposable nappy will absorb 15 times its weight in water. This phenomenal absorption capacity is due to the absorbent pad found in the core of the nappy. This pad is composed of two essential elements, a hydrophilic polymer and a fibrous material such as wood pulp. The polymer (eg polyacrylamide) is made of fine particles of an acrylic acid derivative, such as sodium acrylate, potassium acrylate, or an alkyl acrylate. These polymeric particles act as tiny sponges that retain many times their weight in water.
An interesting EEI would be to measure the water absorption properties of the acrylate polymer using 'fake' urine (water, sodium chloride, urea, hydrochloric acid perhaps) in water in appropriate amounts. Does the nappy work equally well on individual solutions of the urine components (or are polar compounds different to non-polar ones)? How does temperature affect its properties? Where to get the polyacrylamide. You could rip open a nappy but a more controlled way would be to buy "water storage crystals" from the hardware shop. Get a Materials Safety Data Sheet (MSDS) for the one you buy to see what percentage of the crystals are polyacrylamide (should be over 90%).
Heat of reaction and E° All chemical and biochemical reactions involve an energy change; e.g., chemical energy may be transferred as electrical, kinetic, light, sound, or (most often) to heat energy. Chemical to heat energy changes occur, for example, in displacement reactions such as: M(s) + Cu2+(aq) → M2+(aq) + Cu(s). Chemical to electrical energy changes occur, for example, in simple electrical cells; thus, a potential difference (V) is observed if the metal (M) is more or less reactive than copper. It would seem reasonable that the amount of heat evolved is directly related to the voltage of the cell. How true is this? Does it hold over a wide range of voltages, and is it concentration dependent? The photo (on the left) below may give you a start but how on earth will you measure the temperature change without heat loss? The photo on the right makes you glad you didn't have one of these on your lap.
Haybale fires - one for farmers
Over the warmer months Rural Fire Services throughout Australia are called to extinguish fires in stored hay as a result spontaneous combustion. These fires are usually the result of a combination of storing hay that is too moist and warmer temperatures. These fires can lead to the loss of valuable feed and stored machinery as well. The phenomena of exploding haystacks has been with us for as long as we have been making hay. Pliny, the Roman Philosopher wrote in 60BC "When the grass is cut it should be turned towards the sun and must never be stacked until it is quite dry. If this last precaution is not carefully taken a kind of vapour will be seen arising from the rick in the morning, and as soon as the sun is up it will ignite to a certainty, and so be consumed". Old microbiology books often contain anecdotal evidence of haystack explosions, one from 1939 states "The stack that sets itself on fire does so in a curious way dependent at first upon both moisture and microorganisms. A really dry stack of hay won't heat spontaneously; a really damp stack can't be set fire to".
Even though hay bales may seem dry, they are actually quite moist. Hay that is considered 'dry' has up to 20% moisture content. Although hay should be stored at about 15% humidity for the best conservation, it is often baled at much higher moisture levels. There are many reasons for this: moist hay has less leaf loss (major nutrients are in the leaves), sun drying reduces its quality, and, most importantly, climatic conditions often do not allow adequate drying.
When hay is bailed and the plant material is either too green or has excess moisture (over 30%, as a result of rain, dew, flood water, etc) it continues to respire, generating heat and water which emerges as a vapour through the leaf pores. Within the confines of the bale, the water condenses and spreads by capillary action. This initiates and promotes fungal and bacterial growth in the bale. These micro organisms produce heat and along with higher external temperatures and humidity, and can reach a peak of 70°C at about 5-7 days. If the relative humidity in the middle of the stack is below 95% then the microorganisms become inactive and the temperature of the stack drops. If the relative humidity in the middle of the stack is above 97% then the resultant heat of vaporisation of the water dissipates the heat rapidly and the temperature of the stack drops. This explains why very wet silage does not explode. However if the narrow window of 95%-97% relative humidity is obtained then the microorganisms continue to produce heat, which cannot escape, which raises the temperature. This temperature rise accelerates the chemical oxidation of the hay releasing more heat quickly raising the temperature within the bale to ignition point (200-280°C) where it will burst into flames.
It all depends on the size and shape of the bale, the moisture content and how tightly it is packed. Even if it doesn't catch on fire the presence of micro-organisms can be a health problem: a disease known as Farmer's lung disease - a pulmonary affection caused by an allergic reaction to the inhalation of these thermophilic actinomycetes and molds. Farmer's lung typically produces shortness of breath, cough, and fever. If not adequately treated, this disease can lead to severe and irreversible lung damage. This suggests a great EEI and one with great social and economic importance.
A traditional method of monitoring hay stack combustion potential used by farmers has been to poke a section of steel rod into bales in the hay stack and leave for several hours before removing it and feeling for any heat build-up. But for an EEI you have to do better than that.
However, if you are using a standard rectangular bale you can just push a thermometer in to it. For a big bale you would need to get a length of metal pipe with holes drilled in it and bash it into the bale - and then lower a thermometer (attached to a string) gently down the pipe until it is centered.
The most common bales used in Australia are the two string bales, which are approximately 900mm long x 450 mm deep x 350 mm high (16 kg). A prime shredded lucerne hay bale costs about $13.
Strength of plastic If you've ever lifted a full plastic shopping bag you'll know that some are stronger than others. Manufacturers make plastic objects with different strengths to suit different needs. It is not only thickness that is important, but the type of plastic, its density and amount of crosslinking. For example PE comes in several types: high-density polyethylene (HDPE), low-density polyethylene (LDPE), medium-density polyethylene (MDPE) and so on.
You could imagine what UHDPE and VLDPE stand for. High-density polyethylene resin has a greater proportion of crystalline regions than low-density polyethylene. The size and size distribution of crystalline regions are determinants of the tensile strength of the end product. HDPE, with fewer branches than MDPE or LDPE, has a greater proportion of crystals, which results in greater density and greater strength. LDPE has a structure with both long and short molecular branches. With a lesser proportion of crystals than HDPE, it has greater flexibility but less strength.
Why not make this an EEI? You could compare tensile strength with density and perhaps use temperature as a second IV. How do PP and PE compare if they have the same density? Hmmm! I'd have a look at the Australian Standard ASTM D 638 Test method for tensile strength of plastics. You don't need a fancy machine - you can do it at school.
Biodegradable Plastics With the concern for plastics on the environment, many manufacturers now provide biodegradable plastics. For example, the plastic wrappers that many magazines and journals come in are biodegradable (see the photo below of the wrappers for Chemistry in Australia, and Australian Physics). It would be interesting to see how biodegradable these plastics are. Is it due to UV light, water, heat or what.
I suspect UV light is a likely candidate but a perusal of a manufacturer's website should reveal the answer. Non-biodegradable bags are also available. You may have a UV light at school or else you can get a UV fluorescent bulb or tube from the hardware store for about $8. This would be an interesting EEI and the strength testing could be based on the suggestion above (or maybe you have a better idea). May take a few weeks to get a reasonable result so get started early.
Steam distillation of eucalyptus oil Eucalyptus oil is used as component in pharmaceutical preparations to relieve the symptoms of influenza and colds, in products like cough sweets, lozenges, and inhalants. It has antibacterial effects on pathogenic bacteria in the respiratory tract. It used to be a big industry in Australia but has declined as cheaper imports have taken over. Nevertheless, eucalyptus oil, olive leaf oil and ti-tree oil are of vital importance to Australian industry - and society.
One of the most disappointing laboratory experiments you can find is the steam distillation of these oils from leaves. They never work very well and you usually end up with a disappointing emulsion - not clear oil. For a good EEI you would need to do more than just extract some oil; you could have a go at improving the method by trialling different heating and collection methods, different aged leaves and so on; all carefully thought out and justified - not just trial-and-error. If you are stuck you could look at oranges or cloves. A trip to an olive leaf distillery would be a fun day out. The photos would look good in your report.
Soapmaking - the saponification of vegetable oil A soap is the sodium or potassium salt of a long chain fatty acid. Soap making has been around for thousands of years and its manufacture is quite simple. However, there are many pitfalls because the chemistry involved is quite complex. A good EEI would be to make soaps from both sodium hydroxide and potassium hydroxide using a variety of saturated and unsaturated vegetable oils and to compare their properties with commercial soaps and detergents.
Instructions for making soap can be found easily but you'd need to work out ways (and reasons) for changing the reactants and their quantities: that is, what problem are you trying to solve, and what is your hypothesis? To keep the investigation manageable, you would be wise to consider just two independent variables (perhaps type of hydroxide and saturation of the oil) and control the rest (salt, temperature, concentrations etc). The tests might involve suds formation in hard and soft water and ability to remove an oil spot. You could add some perfume and give the leftovers to mum for Mother's Day.
Breaking strain and crosslinking in polymers
Electrolysis of solutions Electrolysis is commercially highly important in the separation of elements from naturally-occurring sources such as ores using an electrolytic cell. It involves the passage of an electric current through an ionic substance that is either molten or dissolved in a suitable solvent, resulting in chemical reactions at the electrodes and separation of materials. It is used in the production of metals such as aluminium, lithium, sodium, potassium and magnesium, and of non-metals such as chlorine. The electrolysis of water produces hydrogen and oxygen and that could make an interesting EEI.
It is known that for water to be electrolysed, it has to have an ionic substance added such as sodium chloride. You could see how the efficiency of the electrolysis is affected by the voltage across the electrodes, and by the concentration of salt present. You'd need to relate your results to the E° value for the non-spontaneous reaction and what happens at voltages lower than that. You may look at the changing rate of generation of the gases as time passes or at the volume after a set time. It's up to you. Does the car ad below make sense? Is it chemically feasible?
Natural buffers Our blood cannot tolerate a drastic shift in pH. It's a good thing, then, that human blood contains a buffer of carbonic acid, H2CO3, and sodium bicarbonate, NaHCO3. This buffer regulates drastic shifts in the pH of our blood. If this buffer system was absent from our blood, the eating acidic or basic foods would cause the pH would swing too high (alkalosis) or too low (acidosis) and the result could be deadly. Another buffer system is that of a mixture of 0.1M Na2HPO4 and 0.1M NaH2PO4. As you add 0.1M NaOH or 0.1M HCl to the buffer solution and record its pH if will be noticeably different to that if you just used water instead of the buffer. Is the buffer any better if you used 0.5M solutions? What if you didn't have equal concentrations? Your EEI could be to locate a buffer system in nature (eg a lake) and test it out using natural environmental chemical changes (eg acid rain, increased greenhouse gases) and find if it has any limits.
Electroplating Electroplating is a common industrial process. It is used to bestow some particular property on an object that it doesn't normally have, for example, abrasion and wear resistance, corrosion protection (galvanising, anodising), or aesthetic qualities (nickel or chrome plating). By applying an electric current, a layer of metal such as copper or nickel can be deposited onto a conductive object. In industry currents of about 500 A are common but in the laboratory a 12V power pack can suffice. A simple experiment that can form the basis of an EEI involves the use of a copper plate and a graphite rod as the cathode and anode, respectively. Nickel ion solution is used as the electrolyte.
Under the influence of the battery, positively charged nickel ion can migrate to the cathode, pickup electrons and deposit on the surface of copper electrode; and there you have nickel plating. You could investigate the role of 'strike': initially, a special plating deposit called a "strike" may be used to form a very thin plating with high quality and good adherence to the substrate. This serves as a foundation for subsequent plating processes. A strike uses a high current density and a bath with a low ion concentration. The process is slow, so more efficient plating processes are used once the desired strike thickness is obtained. The striking method is also used in combination with the plating of different metals.
Or you could investigate current density (amperage of the electroplating current divided by the surface area of the part) in this process strongly influences the deposition rate, plating adherence, and plating quality. The higher the current density, the faster the deposition rate will be, although you get poor adhesion. You may even produce some nice jewellery for mum.
Crosslinking in Slime Slime is merely polyvinyl alcohol (PVA) that has been crosslinked by the addition of borax Na2B4O7.10H2O (sodium tetraborate). Various types of slime have been manufactured but the polymer polyvinyl alcohol is reasonably cheap and is readily available from suppliers because it is widely used as a thickener, stabiliser and binder in cosmetics, paper cloth, films, cements and mortars. Crosslinked PVA is used in hot or cold packs as they are not dangerous if the fluid leaks out. pH is critical in maintaining the crosslinks in slime. Too much acid will weaken the gel but this can be restored with the addition of alkali.
A good EEI would be to test the resultant viscosity (you design the apparatus and procedure) as increasing amounts of borax is added (but you must hypothesise and theorise first); and/or to increase the [H+] by the addition of acid and then lowering it by the addition of NaOH. Another great EEI.
Electrorefining metals Virtually all copper produced from ore receives an electrolytic treatment by electrorefining from impure anodes. In electrorefining, the anodes consist of unrefined impure metal, and as the current passes through the acidic electrolyte the anodes are corroded into the solution so that the electroplating process deposits refined pure metal onto the cathodes. In order to achieve high production rates, high current densities are desirable but an excessive current density causes at least two problems: increased impurity levels in the cathode deposit; and anode passivity occurs at current densities above 25-28 mA/cm2. Hence, in industry, the current density is always low. An interesting experiment would be to set up an electrorefining cell for copper and find out the optimum current density and/or acid concentration.
Aspirin hydrolysis using a spectrometer Aspirin is the common name for acetyl salicylic acid (ASA) and is an important drug on the market today. For example, the treatment of thromboembolism often requires the use of ASA. Aspirin is rapidly absorbed from aqueous solution and hydrolysis occurs during the absorption phase and first pass through the liver. It then is converted to salicylic acid (SA) in the blood, predominantly in the liver but also in blood cells, plasma, and kidneys.
The hydrolysis of ASA to SA has been the subject of many investigations and can be studied in a high school laboratory if equipped with a visible spectrometer such as a Spectronic 20. The rate constant "k" for the reaction depends on pH, temperature, buffer concentration, and ionic strength. It can be followed by measuring spectrophotometrically the appearance of the complex of SA with ferric chloride, FeCl3. The method can be found in The Journal of Chemical Education 2000, Vol 7, p 354 by L. Borer and E. Barry.
Conductivity of solutions Electrical conductance is important in a variety of scientific contexts; e.g., nerve impulses, electroplating, electrical cells, and the extraction of metals by electrolytic reduction. You might expect the conductance of an aqueous ionic compound to be dependent on several independent variables, including the concentration of dissolved compound. A good EEI would be to examine this hypothesis: As the concentration (M) of sodium chloride increases, within the range ?? to ?? mol/L, the conductances (C) of the aqueous solutions increase in direct proportion. However, does the relationship hold for all concentrations, temperatures, electrode area, electrode separation and voltages?
Ion exchange resin Ion-exchange resins are widely used in different separation, purification, and decontamination processes. The most common examples are water softening and water purification. Most recently, they can be used for biodiesel recovery. In many cases ion-exchange resins were introduced in such processes as a more flexible alternative to the use of natural or artificial zeolites. The resins are usually small plastic beads that contain ionic groups attached to a polymer-based resin. These ionic groups can be exchanged for similarly charged ions. There are many possibilities for an EEI here.
Start with a cation exchange resin and plan an experiment to find the extent to which Na+ ions (from say NaCl solution) exchange with the hydrogen ions on the resin. You could Investigate the rate of exchange of ions by leaving the exchange resin in the sodium chloride solution for different periods of time (and plot graphs). Or you could investigate the effect of using different concentrations of sodium ions on the rate of exchange or the effect using cations such as potassium, calcium, aluminium, copper (II), and iron (II). Do they exchange to the same extent and at a similar rate?
V
Concrete hydration The importance of concrete in modern society cannot be overestimated. Look around you and you will find concrete structures everywhere such as buildings, roads, bridges, and dams. There is no escaping the impact concrete makes on your everyday life. Concrete is prepared by mixing cement, water, and aggregate together to make a workable paste. It is molded or placed as desired, consolidated, and then left to harden. Adding gypsum, CaSO4, to Portland cement prolongs the hardening. The most important compounds present in cement are: 3CaO•Al2O3, tricalcium aluminate; 3CaO•SiO3, tricalcium silicate; 2CaO•SiO3, dicalcium silicate; and CaO, calcium oxide. The 2CaO•SiO3 reacts slowly with water to yield Ca(OH)2 and H2SiO3. This reaction not only helps in holding the material together, but also makes the concrete less pervious to water.
The hardening process is due in part to the hydration of the compounds present and is probably influenced by the crystallization of these hydrates. Concrete with too little water may be dry but is not fully reacted. The properties of such a concrete would be less than that of a wet concrete. You could make up thin slabs of concrete in a shallow trough with different amounts of water and test their breaking strain. What if you were unable to get fresh water - would seawater be just as good? If you try other additives, you have to say why you think they'd work (otherwise it's not chemistry - it's just backyard trial-and-error). The possibilities are endless.
Recycling an aluminium can Recycling an aluminium can save 95% of the energy required to produce a new can from ore. Aluminum cans are easily recycled into new aluminum cans, but they can also be recycled into other useful aluminum products. A good EEI would be to convert aluminum cans into alum (potassium aluminum sulfate). Large amounts of alum is used by the paper industry as a filler in paper and secondly for drinking water purification. Merely converting can to alum is hardly the basis for an EEI. You'd need to apply some problem-solving and creative thinking. Perhaps you could look at ways of maximizing the yields and minimizing the input energy (heat) and chemical resources (KOH).
The health of our river The Brisbane River and the waterways of the Moreton Bay catchment play a vital role in the economy, lifestyle and liveability of South-East Queensland. These waterways support the largest population of any catchment in the State and provide a nationally significant drinking supply. They also provide recreational and employment opportunities and are of cultural significance to the people of the region. But they are under enormous pressure from population growth. Scientific research indicates current levels of human impact on our waterways are unsustainable and our behaviour and practices must change if we are to halt and reverse the current decline in water quality.
This context affords some great EEIs but you need to be careful that you don't just end up testing water samples and making some statements about water quality. If you plan to assess the health of the river you would need to state which tests you are using, why, the techniques, the sampling, the appropriateness of the tests if the water is saline, and so on. A good EEI would also be to ask "What is the effect of depth of water and temperature on dissolved oxygen as measured by using the Winkler technique?" or "What is the effect of salinity on chemical tests?" or "What is the more effective way of measuring salinity and what is the effect of tides on salinity?". Many schools use this context for EEIs and its societal importance is obvious.
Strength of fired pottery clay Pottery is one of the oldest human technologies and art-forms, and remains a major industry today. It is made by forming a clay body into objects of a required shape and heating them to high temperatures in a kiln to induce reactions that lead to permanent changes, including increasing their strength and hardening and setting their shape. Firing produces irreversible chemical changes in the body. As a rough guide, firing temperatures are in the range of about 1000 to 1400°C. However, the way that ceramics mature in the kiln is influenced not only by the peak temperature achieved, but also by the duration of the period of firing. A good EEI (especially if you do Senior Art) might be to examine the hardness of the fired clay as a function of temperature; or as a function of time. If you were more adventurous you could look at different atmospheres within the kiln. One word of caution. This is a chemistry EEI and chemistry must be at its heart to distinguish it from applied technology or art. A pyrometric cone (see photo below) is a spike-shaped piece of clay used to measure temperature in a kiln when firing pottery. Cones have carefully calibrated melting points, indicated by their cone number. They are used to visually determine when a kiln has reached a desired temperature, by observing when a given cone in an observation port starts to droop. They are very attractive too.
Polarisation of light in acidified sugar solution Certain materials (sugar in this experiment) are optically active. When polarized light passes through an optically active material, its direction of polarization is rotated. The angle of rotation depends on the thickness of the material and the wavelength of the light. You could make up a solution of sugar (the disaccharide called sucrose) and hydrolyse it using dilute acid to form the monosaccharides glucose and fructose:
C12H22O11(sucrose) + H2O + H+ => C6H12O6(fructose) + C6H12O6(glucose) + H+. The product is called invert sugar.
As the reaction proceeds, the degree of polarisation changes and this can be observed using crossed polarisers either side of the solution placed on an OHP. Inverted syrups are sweeter than sucrose solutions and because there is glucose present in inverted sugar syrup it is substantially more hygroscopic (water retaining) than sucrose. This means that the syrup tends to keep products made with it moist for longer than when sucrose is used alone. It is likewise less prone to crystallisation and therefore valued especially by bakers. You could look at the effect of angle vs. concentration vs time; depth effects; acidity effects; temperature. If you are after a real challenge you could investigate if the reaction rate constant is dependant on acid concentration.
This suggests a good EEI. Basically, you need to add chips of dry ice of different masses to water in a sealed container and measure the pH. Safety is of the utmost concern here as the dry ice can easily burn you. The big problem is how to control the controlled variables. You may also want to compare distilled water with salt water because the chemical components of the ocean acts as a buffer absorbing more carbon dioxide than freshwater can without a change in pH. Because the temperature of the ocean is also rising from global warming, the temperature variable would be worth considering.
Distillation of alcohol Distillation is one of the oldest and still most common methods for both the purification and the identification of organic liquids. It is a physical process used to separate chemicals from a mixture by the difference in how easily they vaporize. Distillation relies on the fact that the vapor above a liquid mixture is richer in the more volatile component in the liquid, the composition being controlled by Raoult's law. Not all mixtures of liquids obey Raoult's law, such mixtures; called azeotropes, mimic the boiling behavior of pure liquids.
These mixtures when present at specific concentrations usually distill at a constant boiling temperature and cannot be separated by distillation. Examples of such mixtures are 95% ethanol-5% water (bp 78.1°C). I think you could make a successful EEI out of an experiment where you distill various ethanol/water combinations and measure the %ethanol in the distillate as a function of time or temperature of the vapour. You would need to consult vapour pressure diagrams.
Annealing Metals are used for many different purposes. Two hundred years ago, the town blacksmith produced nails, hammers, wheel rims, knives, and horseshoes from the same basic metal. In some applications, a metal must be able to bend easily without breaking, whereas in other cases the metal must resist bending. Today metallurgists can produce these results by using different metals, alloying metals, and by heat treating metals. The substitution of a different metal or using a special alloy is often costly. Therefore heat treatment of a common metal is often the most cost efficient method of producing a metal that has the properties required in a specific application.
Most metals respond to heat treatment, but the treatment temperatures are unique for different metals. A great EEI is to examine the effects of annealing, quenching, and tempering on metals. A steel bobby pin would be a useful starting point but you'd need to control the amount of heating and quenching and see how the properties vary with changes. Ask yourself "what type of treatment produces the hardest metal; and the strongest metal"? The male Blood Elf (below) from the World of Warcraft is carrying a quenching bucket. Nothing to do with chemistry however.
Anthocyanins in wine Anthocyanin pigments are responsible for the attractive red to purple to blue colors of many fruits and vegetables including dark wine grapes. Interest in the anthocyanin content of foods has also intensified because of their possible health benefits. They may play a role in reduction of coronary heart disease, increased visual acuity, as well as antioxidant and anticancer properties. Anthocyanins are relatively unstable and often undergo degradative reactions during processing and storage. Measurement of total anthocyanin pigment content along with indices for the degradation of these pigments are very useful in assessing the color quality of these foods. There is a method used for determining anthocyanins in wine. It was developed by Fuleki and Francis and you'll find it on the web. You'll also need a visible spectrophotometer (520 nm).
Corrosion of roofing steel Corrugated galvanised iron is a building material composed of sheets of hot-dip galvanised mild steel, cold-rolled to produce a linear corrugated pattern in them. Galvanized metals prevent rust not only by protecting the metal from direct oxygen contact, but also by the electrochemistry of zinc. When iron rusts its oxidation state is increased as electrons are transferred away from the metal. Zinc acts as an electron donor in a slightly complex electrochemical reaction, thereby preventing the oxidation of the underlying metal.
Nevertheless, rusting will be inevitable, especially if the local rainfall is at all acidic in nature. So for example, corrugated iron sheet roofing will start to degrade within a few years despite the protective action of the zinc coating. An EEI might be is to see how far the zinc protection extends over bare metal. Small scratches don't rust but if the scratch is 10 mm wide will it? What if kept in a humid atmosphere? Is BlueScope zincalume better than ordinary galvanising?
Paper chromatography of leaves Paper chromatography is an analytical chemistry technique for separating and identifying mixtures that are or can be coloured, especially pigments. This can also be used in secondary or primary colours in ink experiments. Most leaves are green due to chlorophyll. This substance is important in photosynthesis (the process by which plants make their food). You have probably done experiments where the different pigments present in a leaf are separated using paper chromatography. However, to make this a good EEI you need to take it further. Which is the optimum solvent (propanone, ethanol, hexane etc) and why (polar, non-polar, low viscosity, high BPt and so on)?
Diffusion of aqueous ions Diffusion is the process by which molecules spread from areas of high concentration, to areas of low concentration. Diffusion occurs in liquids but more slowly than in gases because the particles are not as free to move about. When a crystal of Pb(NO3)2 and a crystal of KI are placed on opposites sides of a petri dish filled with water a yellow line of PbI2 forms across the dish closer to one crystal than the other.
This gives you an idea of the rate of diffusion of ions. You could repeat this with different combinations so long as they form a precipitate. Is it just the molar mass of the ion, or is it related to the charge, or perhaps something to do with electronegativity. Would anything happen with a non-polar solvent such as hexane, and if not, why not? A great EEI that will keep you entertained for weeks. There will be safety issues with heavy metal ions (eg Pb2+) so be warned.
Migration of ions Ions, being charged, will migrate towards electrodes of opposite charge. For example, the migration of manganate ions (MnO42-) can be observed if you cut a piece of filter paper slightly smaller than a microscope slide and moisten the filter paper with tap water. Then fasten the paper to the slide with crocodile clips and put a small crystal of potassium manganate (K2MnO4) in the centre of the paper. When you connect the clips to a power supply set at 12 V DC you should notice the migration of the coloured manganate ion towards the negative electrode. Occasionally permanganate (MnO4-) and manganate (MnO42-) salts are confused, but they behave quite differently. How different is the speed of migration for larger ions, for ions of different charge. Is voltage related to migration speed. What a great EEI this is turning out to be.
Testing water hardness Tap water in some parts of the country is very pure and is said to be 'soft'. It easily makes a lather with soap. Water from other parts may contain various dissolved impurities and is described as 'hard' water. Temporary hardness may be removed by boiling, but permanent hardness survives the boiling process. You can measure water hardness by finding out the volume of a soap solution (of known concentration, eg 10 g of plain laundry soap per 100 mL of 80 % ethanol or metho) required to form a permanent lather with a known volume of the water to be tested (eg 5 mL) in a test tube.
Actually, this is the standard Clarke's soap solution invented by Dr. Thomas Clarke, Professor of Chemistry at Aberdeen University, in 1843. A interesting EEI would be to see if the amount of soap needed is correlated with the concentration of various ions responsible for hardness (Ca, Mg). You could make up solutions with a range of concentrations of 'hardness' ions and see how much soap is needed to make a permanent lather (one that lasts for 30 seconds) is obtained when shaken. Try adding dropwise increments of the soap solution from a burette. 'Temporary' hard water can be made by using decanting a saturated solution of Ca(OH)2; and permanent hard water can be made by using either 1 g CaSO4•2H2O or 1 g MgSO4•7H2O in 100 mL water.
Permanent hard water contains Ca or Mg salts other than the hydrogen carbonates. Some tests you could do are: untreated deionized water (control), untreated tap water (real life); a comparison of untreated temporary hard water and untreated permanent hard water with boiled temporary hard water and boiled permanent hard water. You could investigate the effect of adding sodium carbonate crystals (washing soda) to temporary hard water; or the addition of adding sodium carbonate crystals (washing soda) to permanent hard water. Analysis of water hardness in major Australian cities by the Australian Water Association shows a range from very soft (Melbourne) to very hard (Adelaide). Total Hardness levels reported in various government reports are listed below:
Return to New Century Senior Physics - textbook Home Page



































































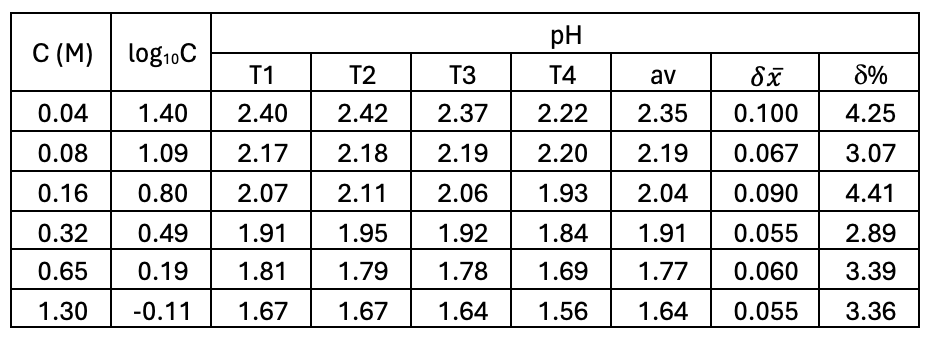
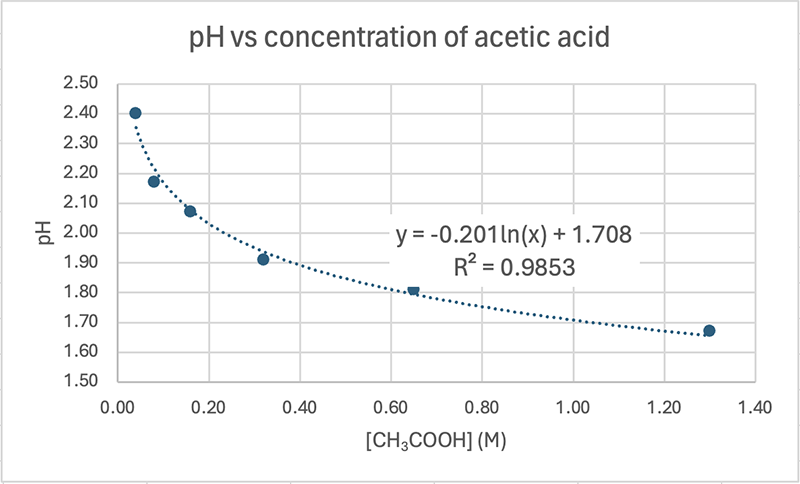
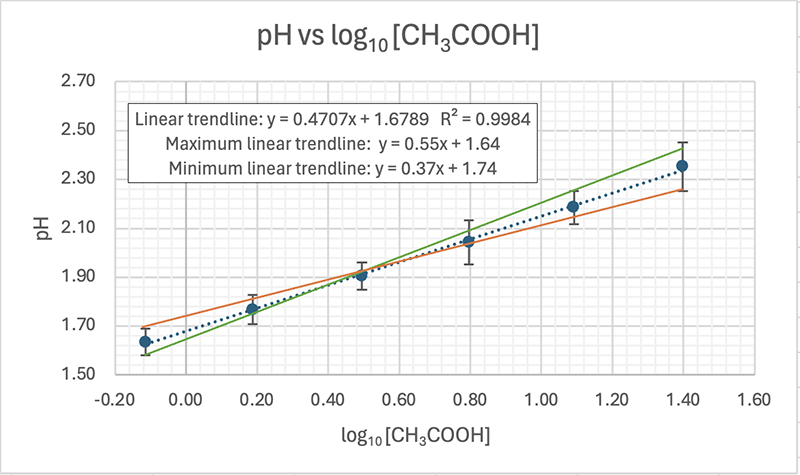
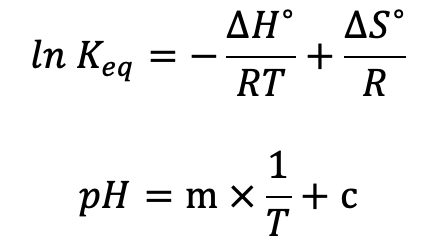

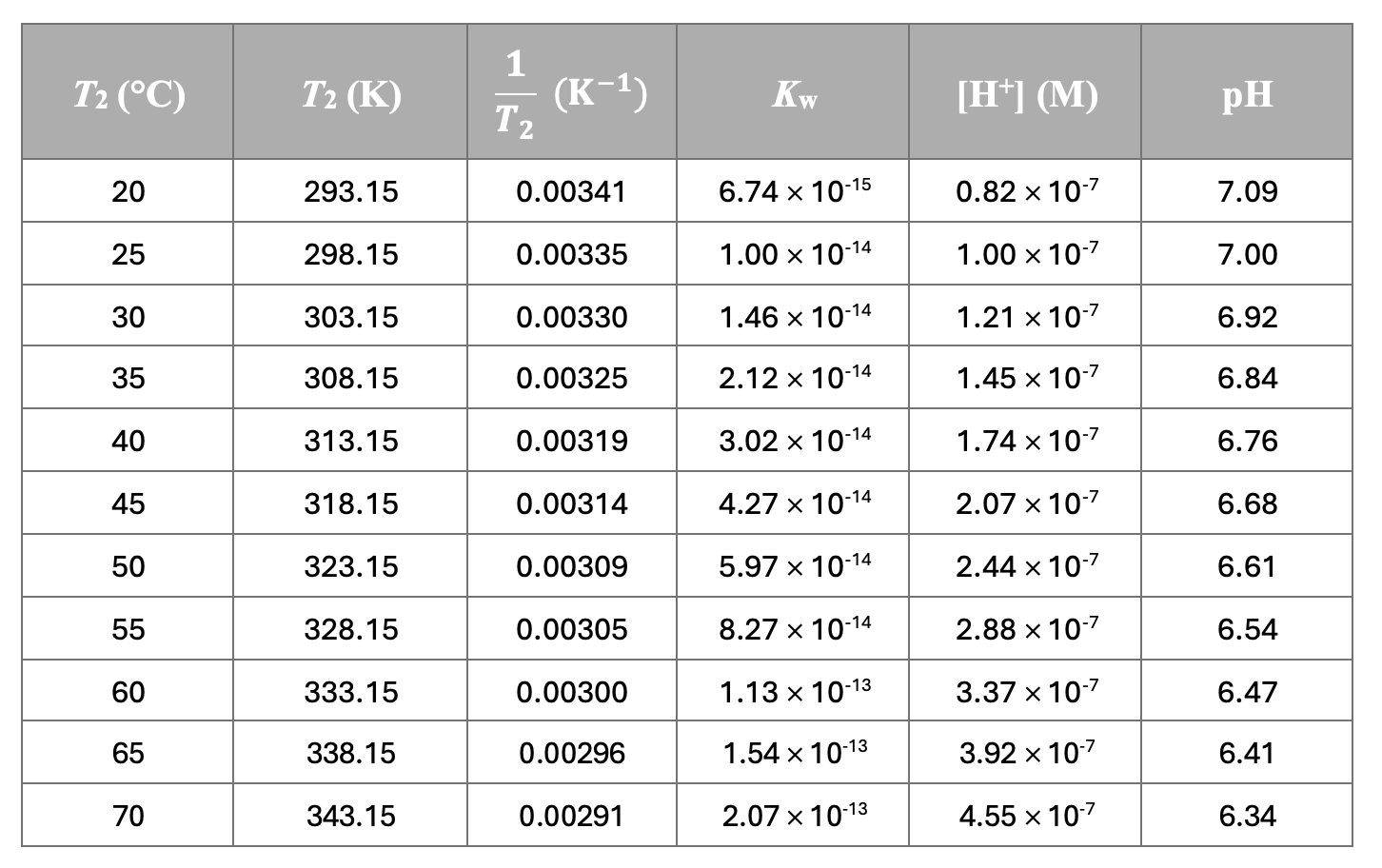
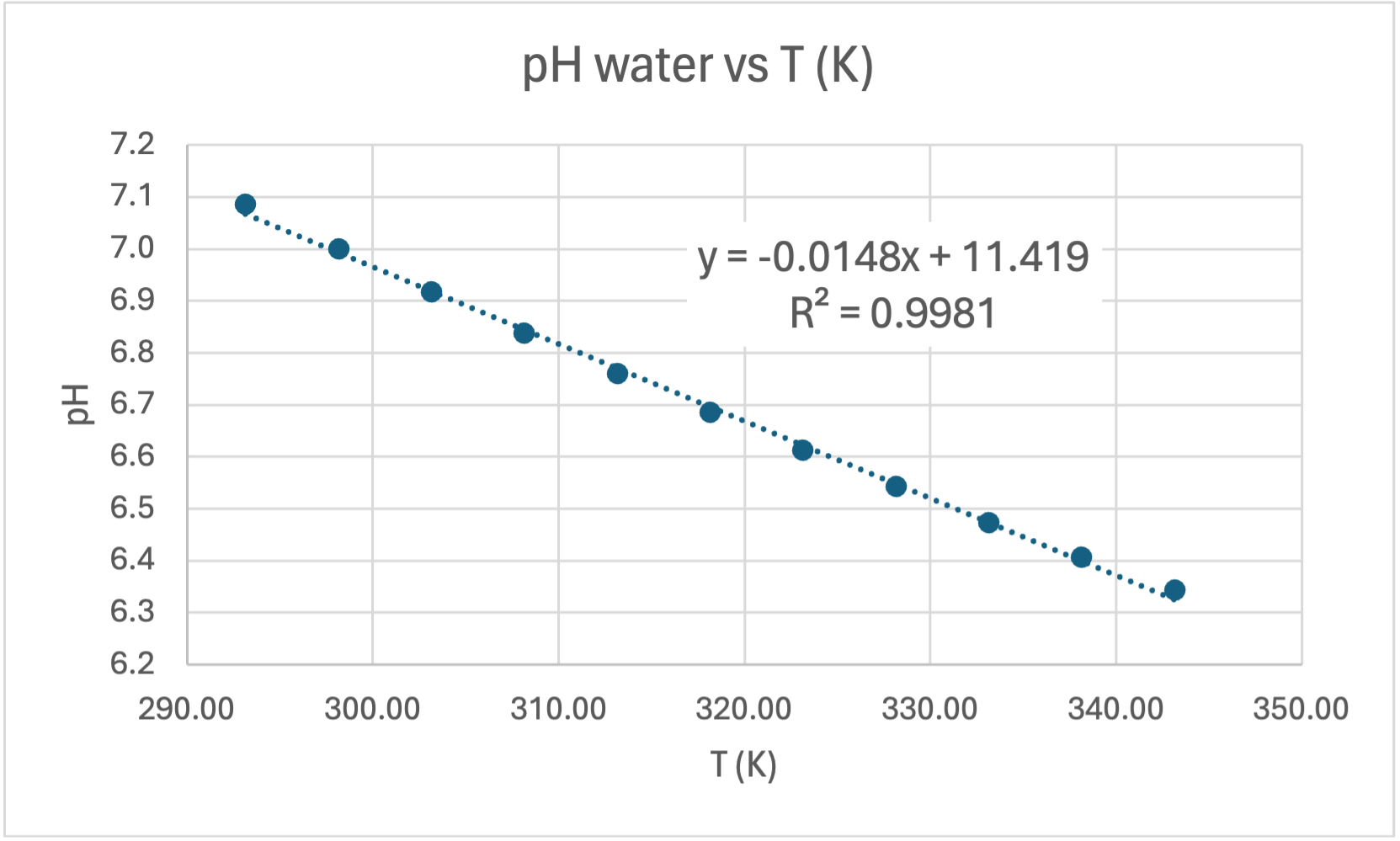
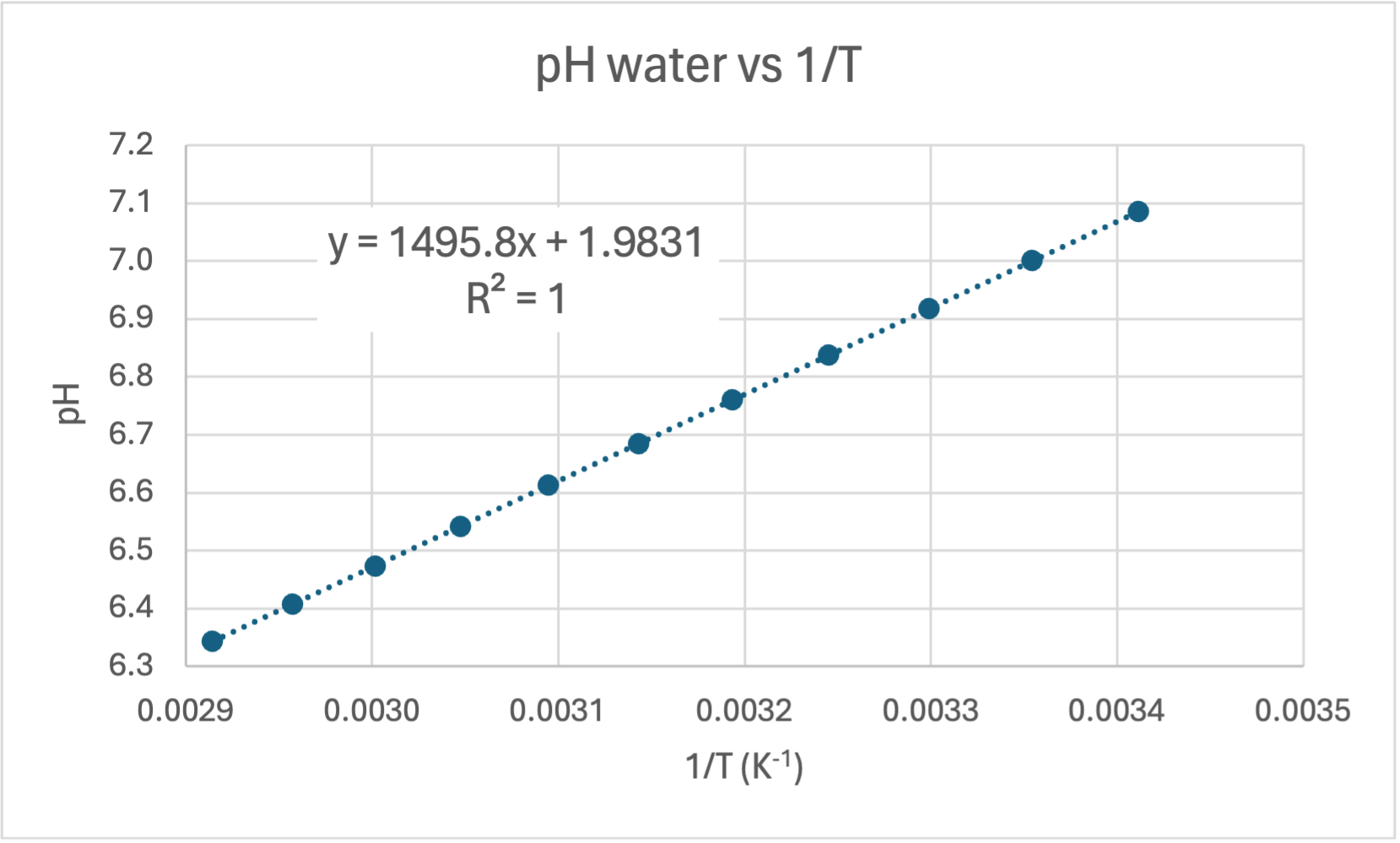


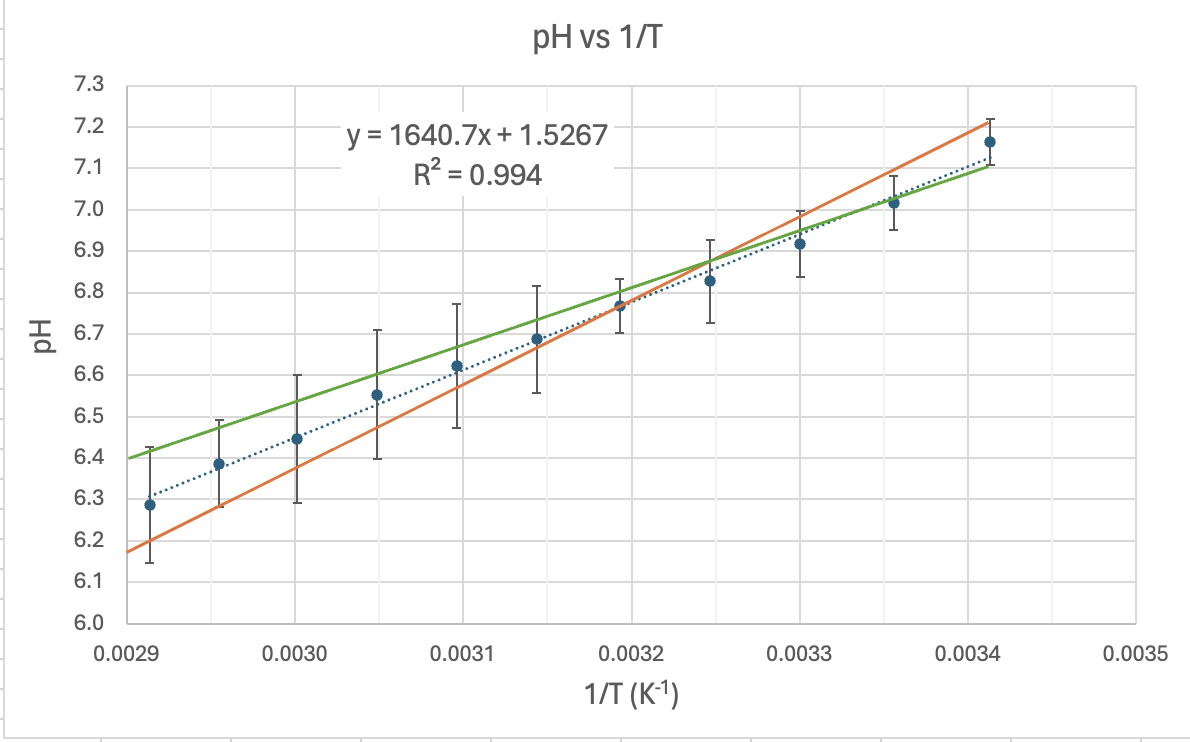
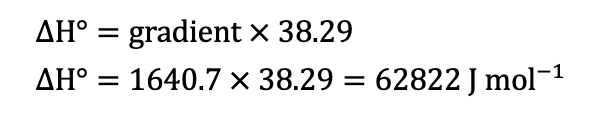
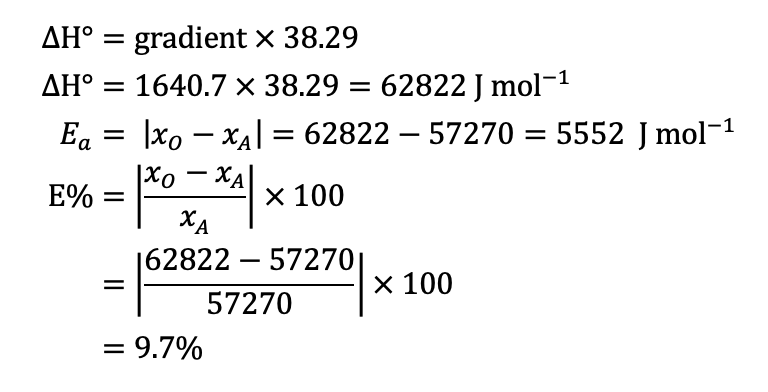

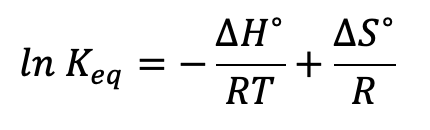


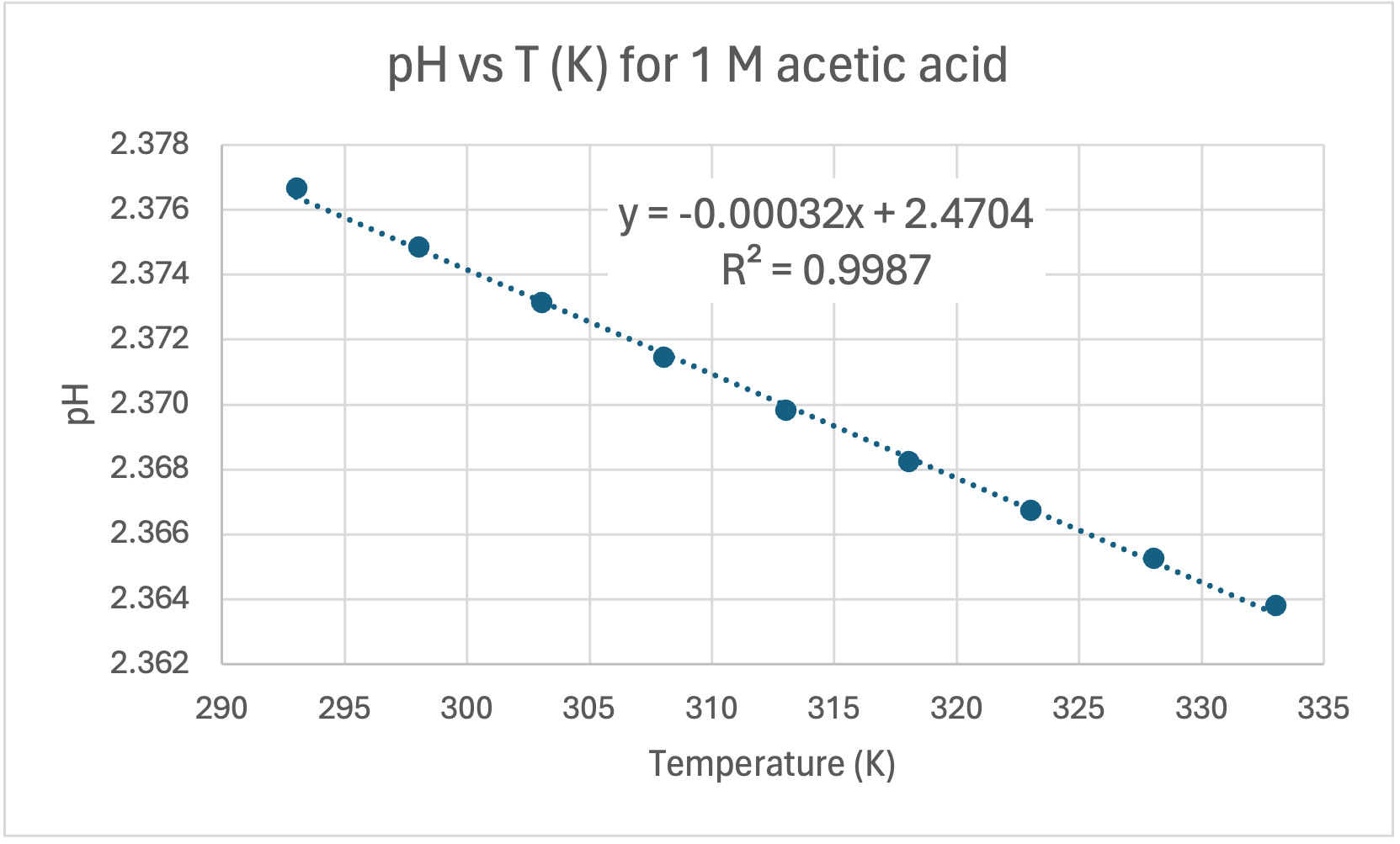
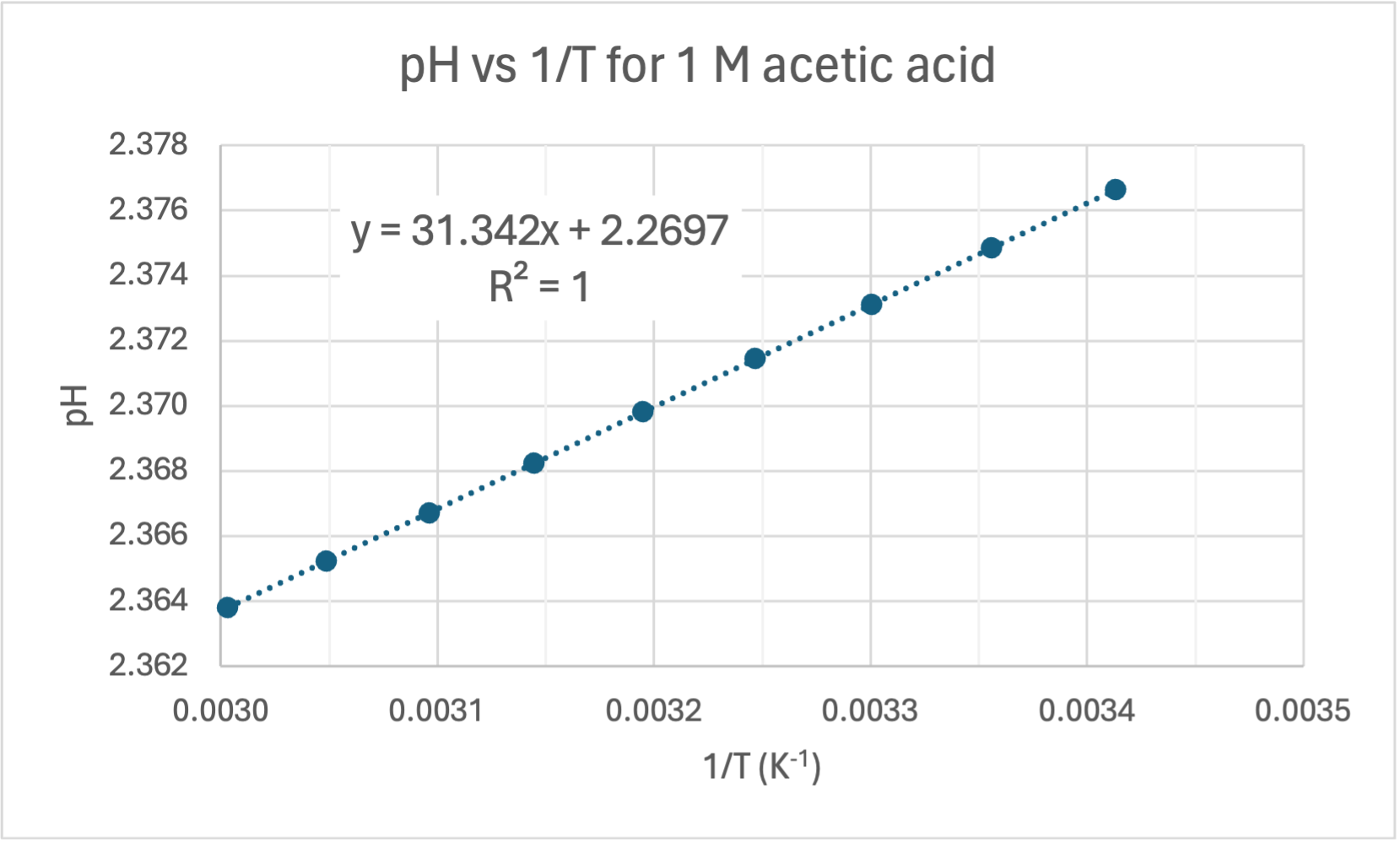


.png)
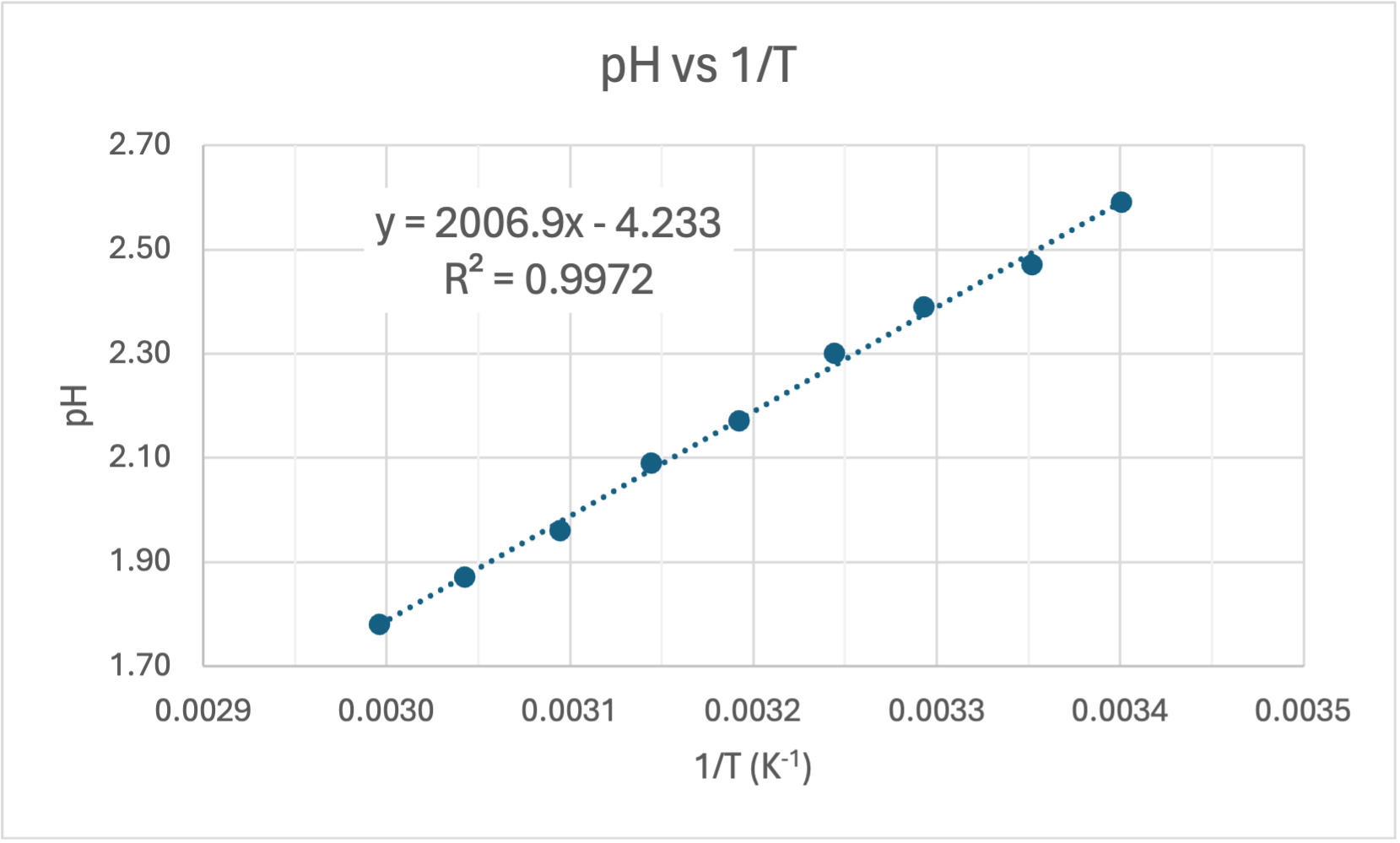


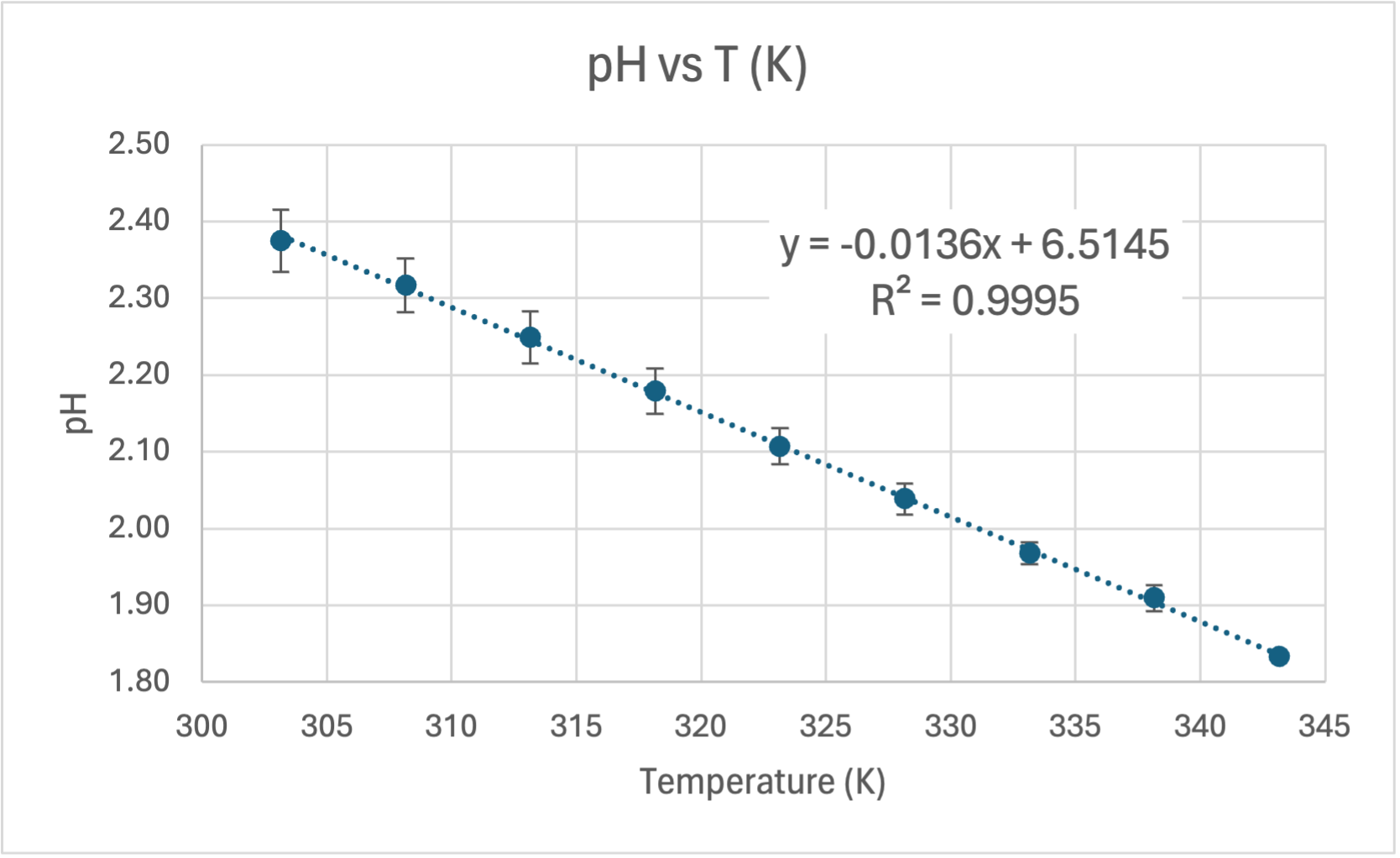
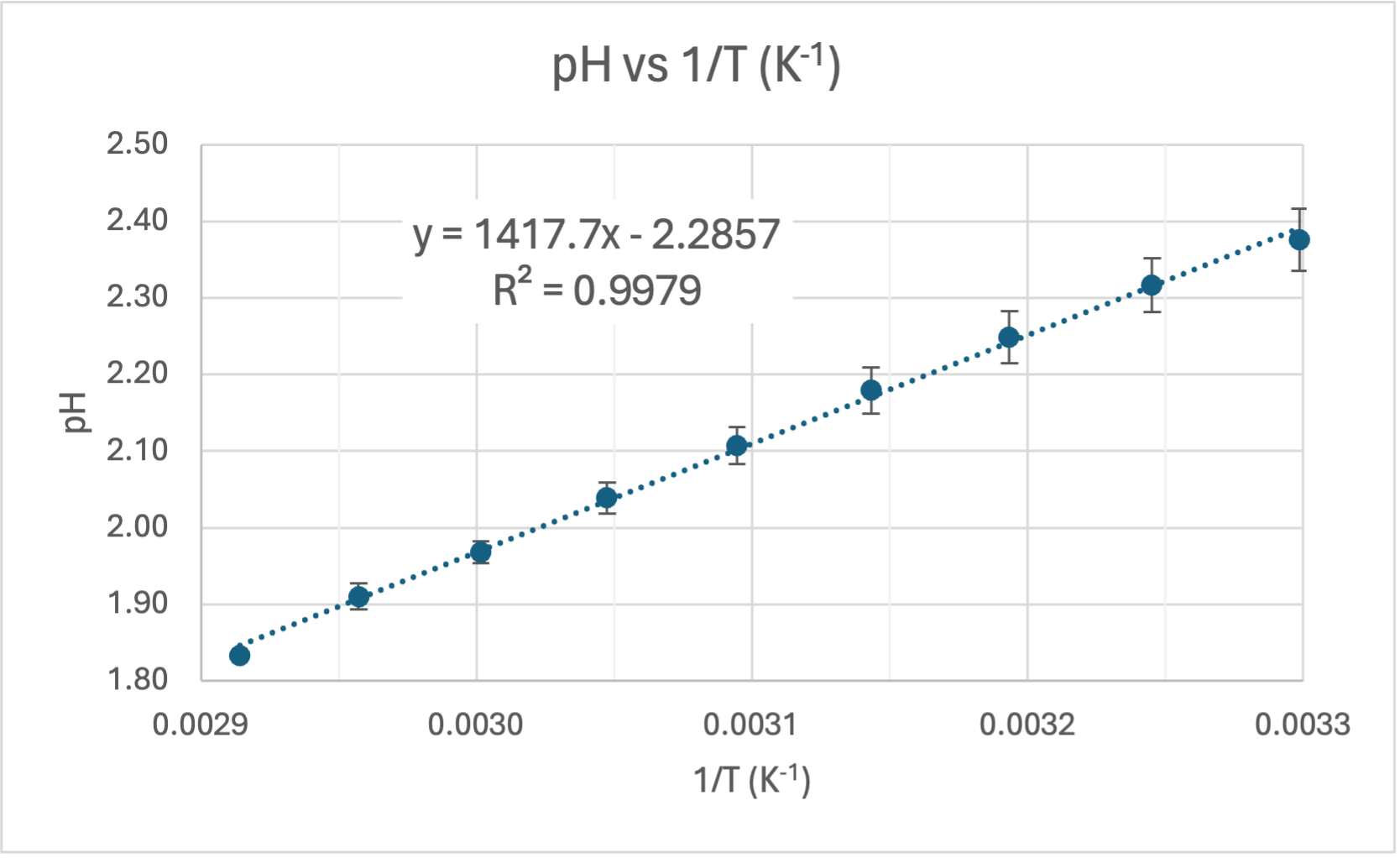
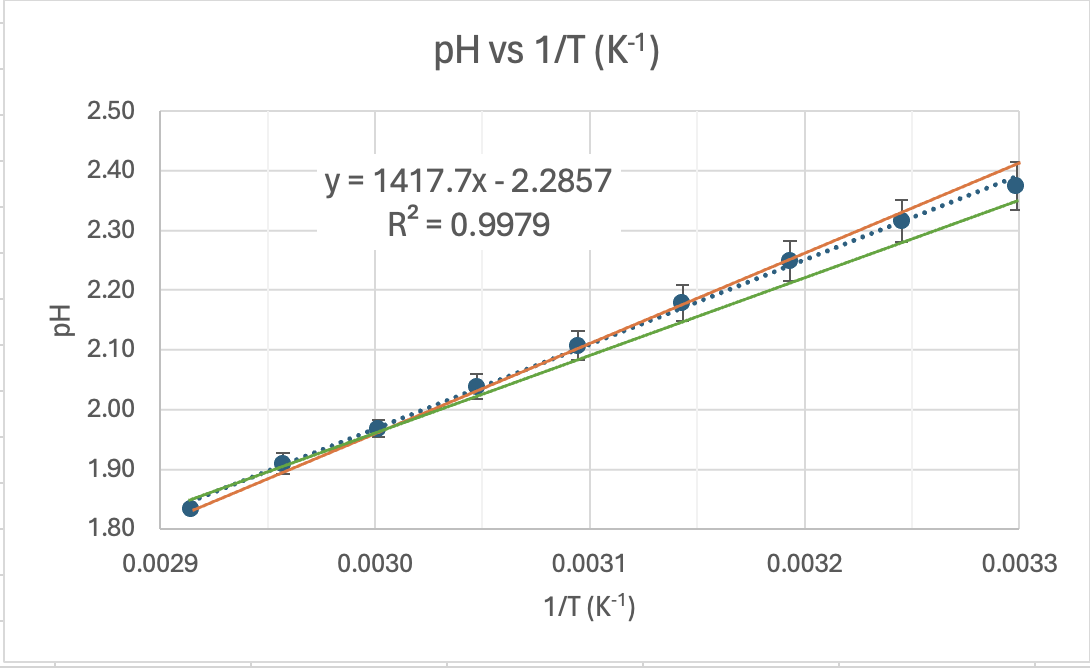






















































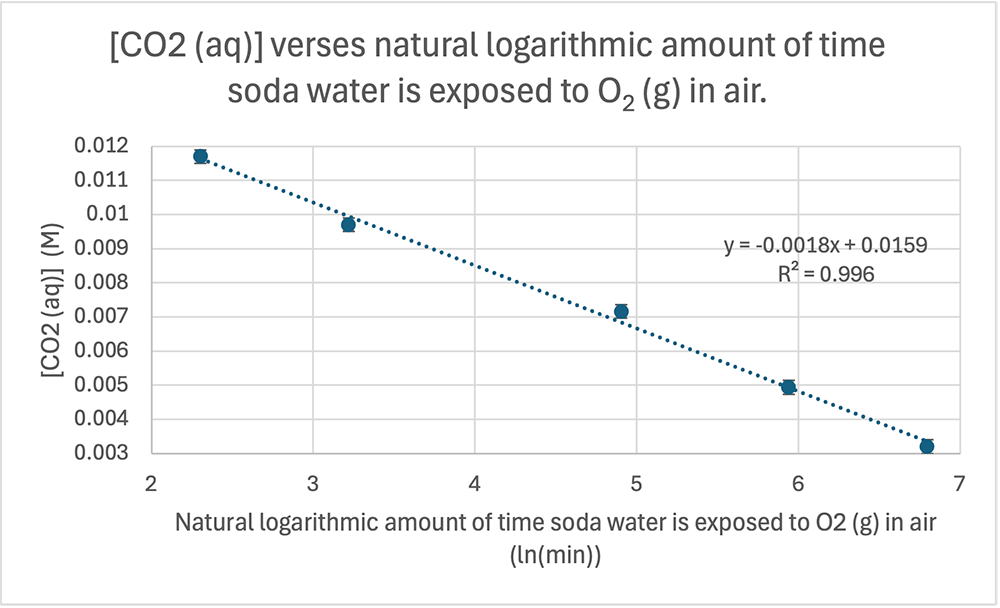
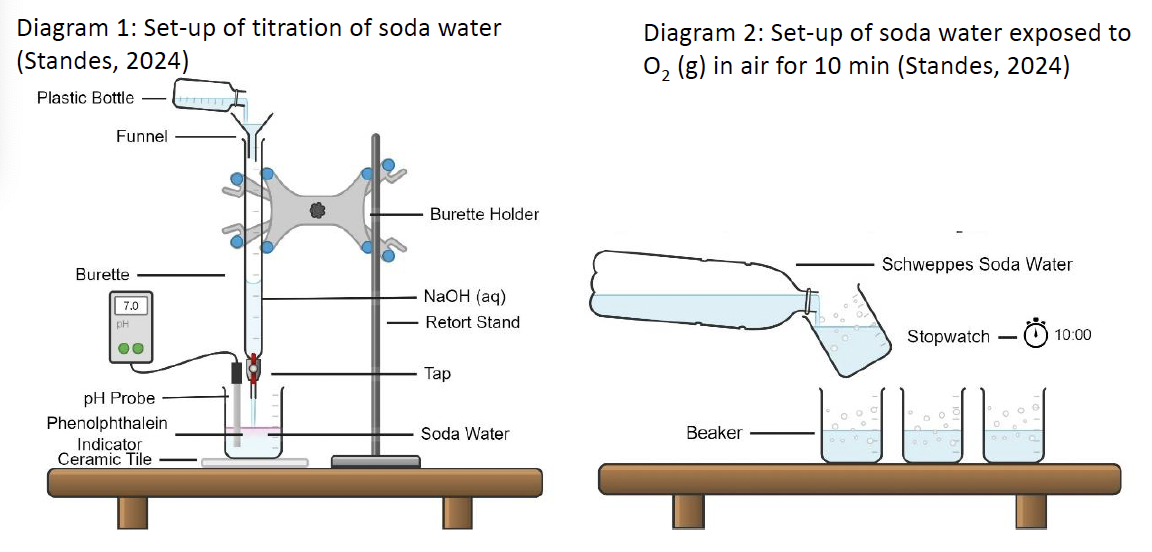







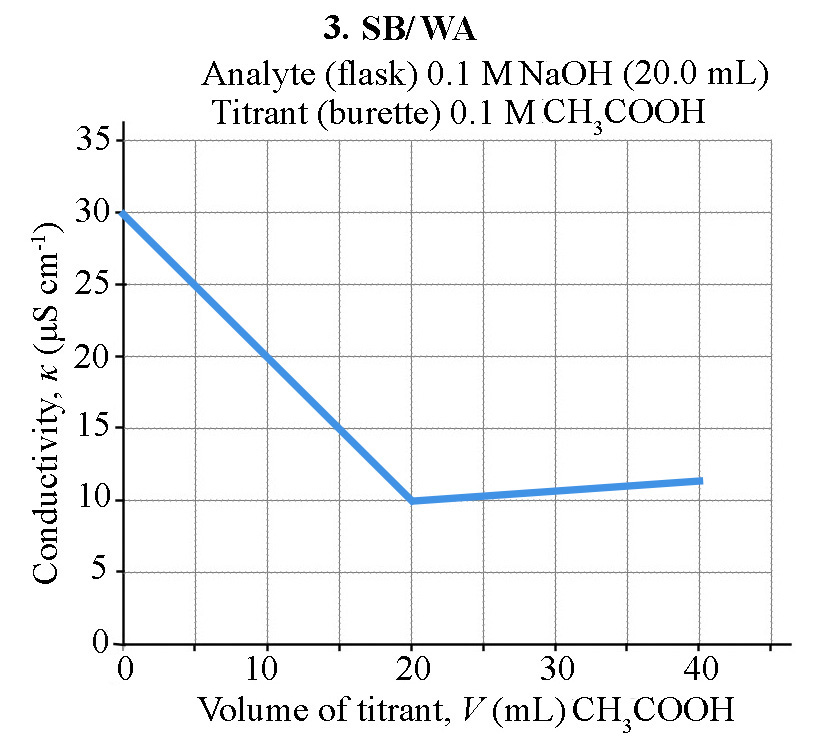
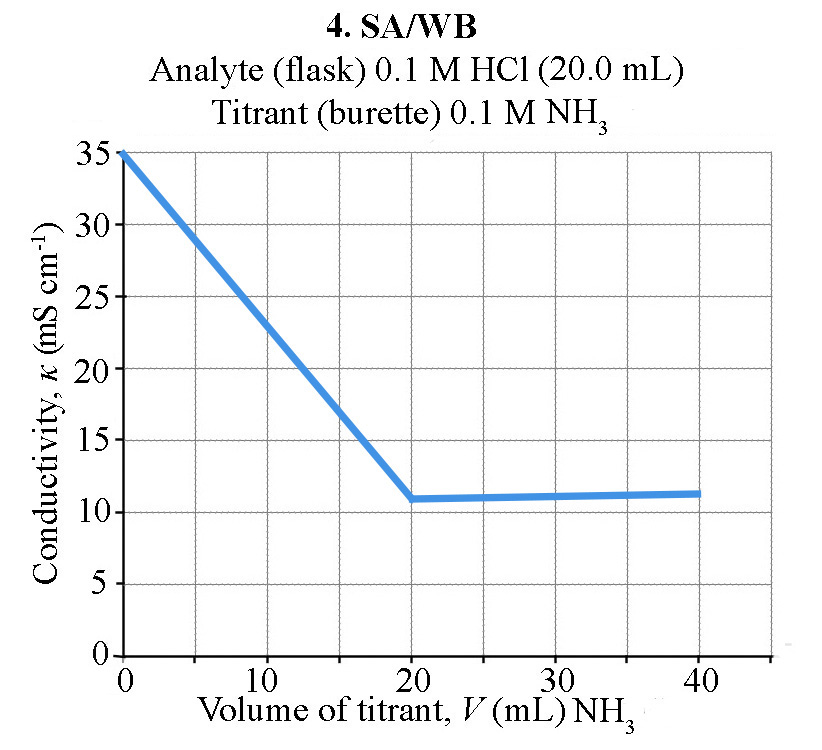
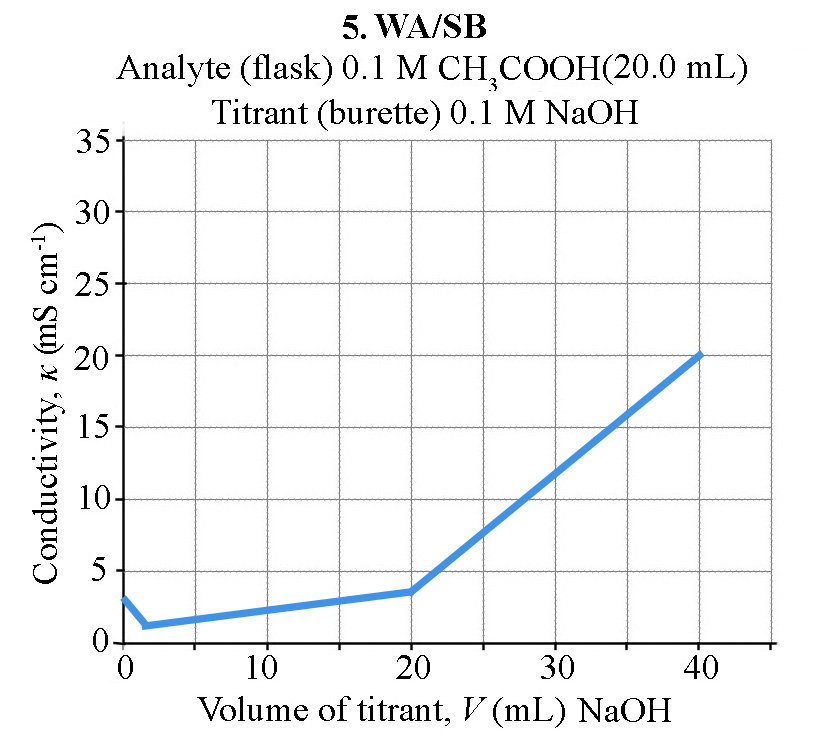
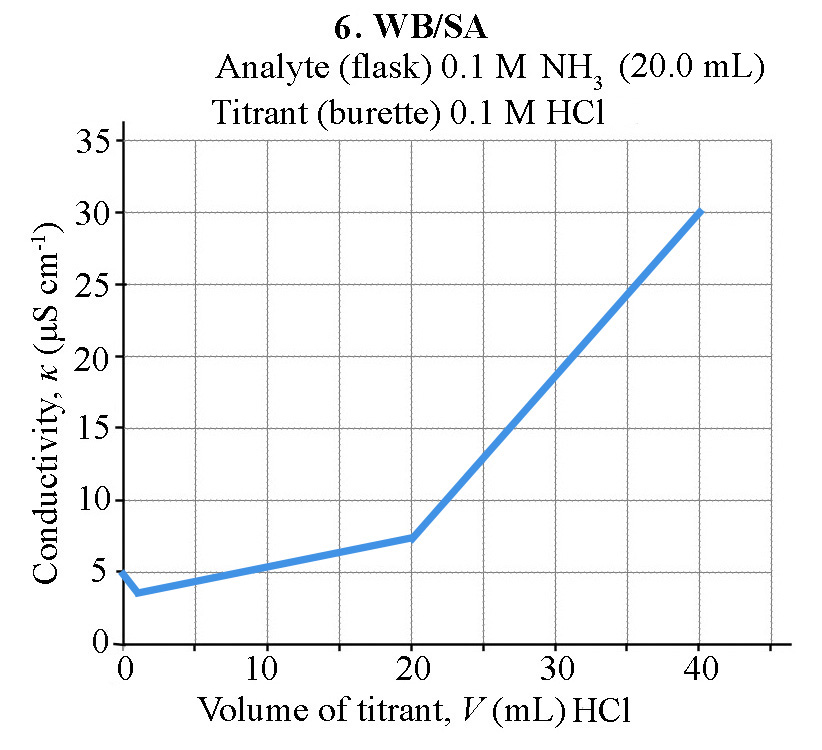
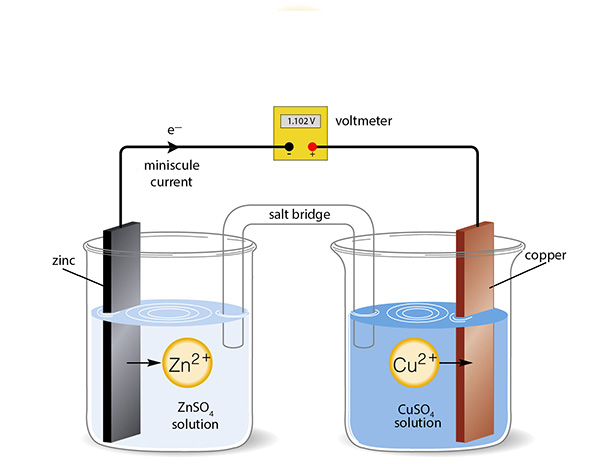































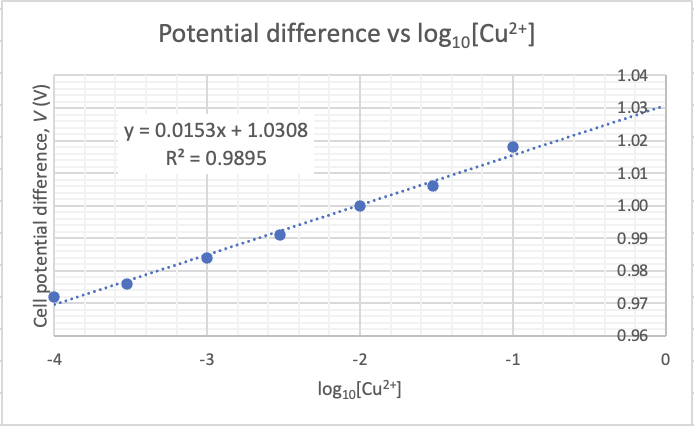
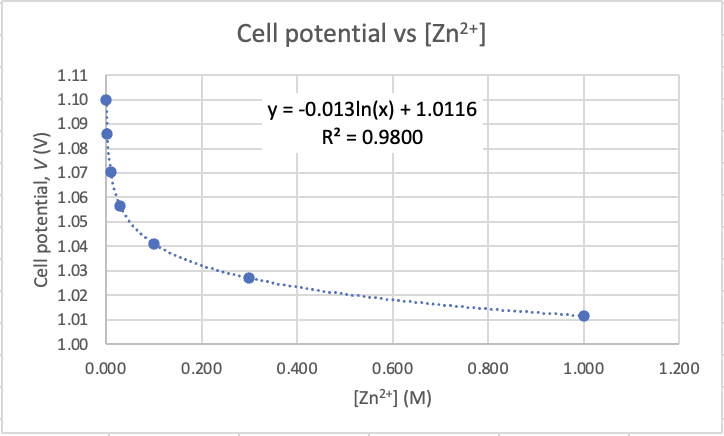
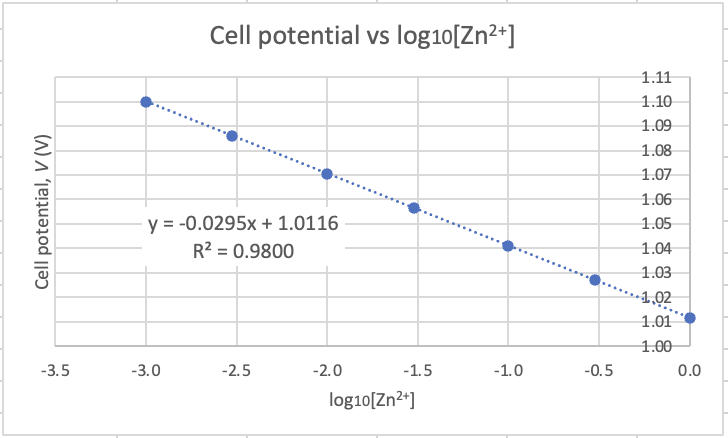








































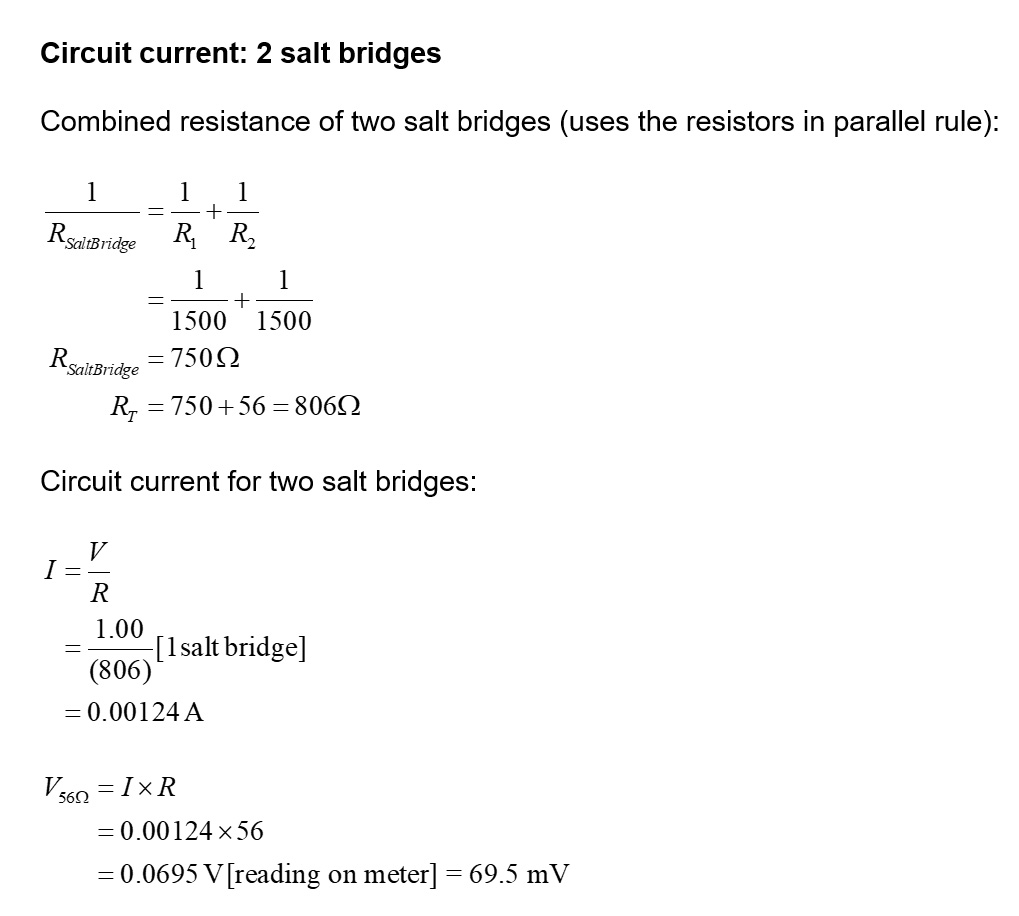




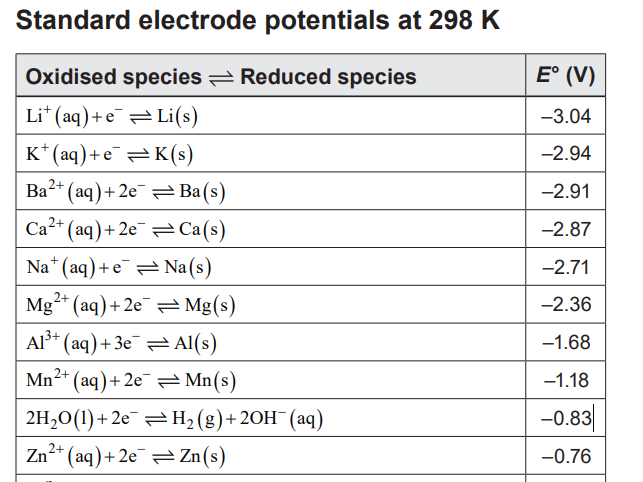






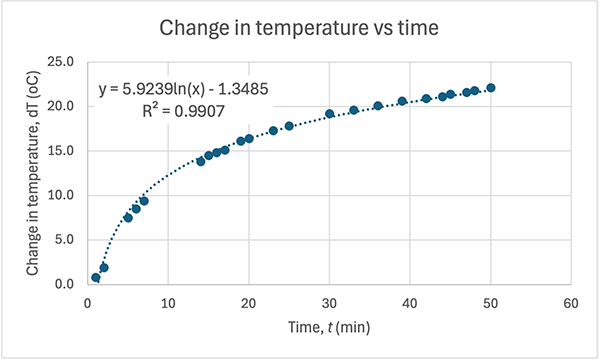
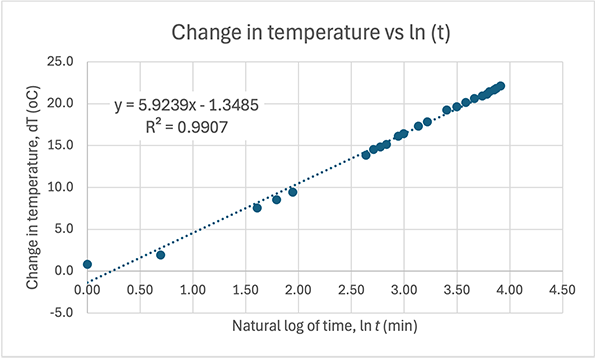


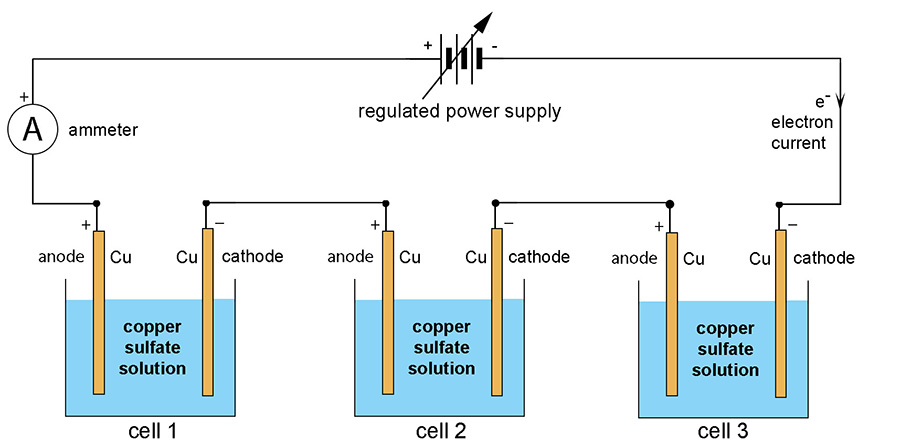




















































































































































































































































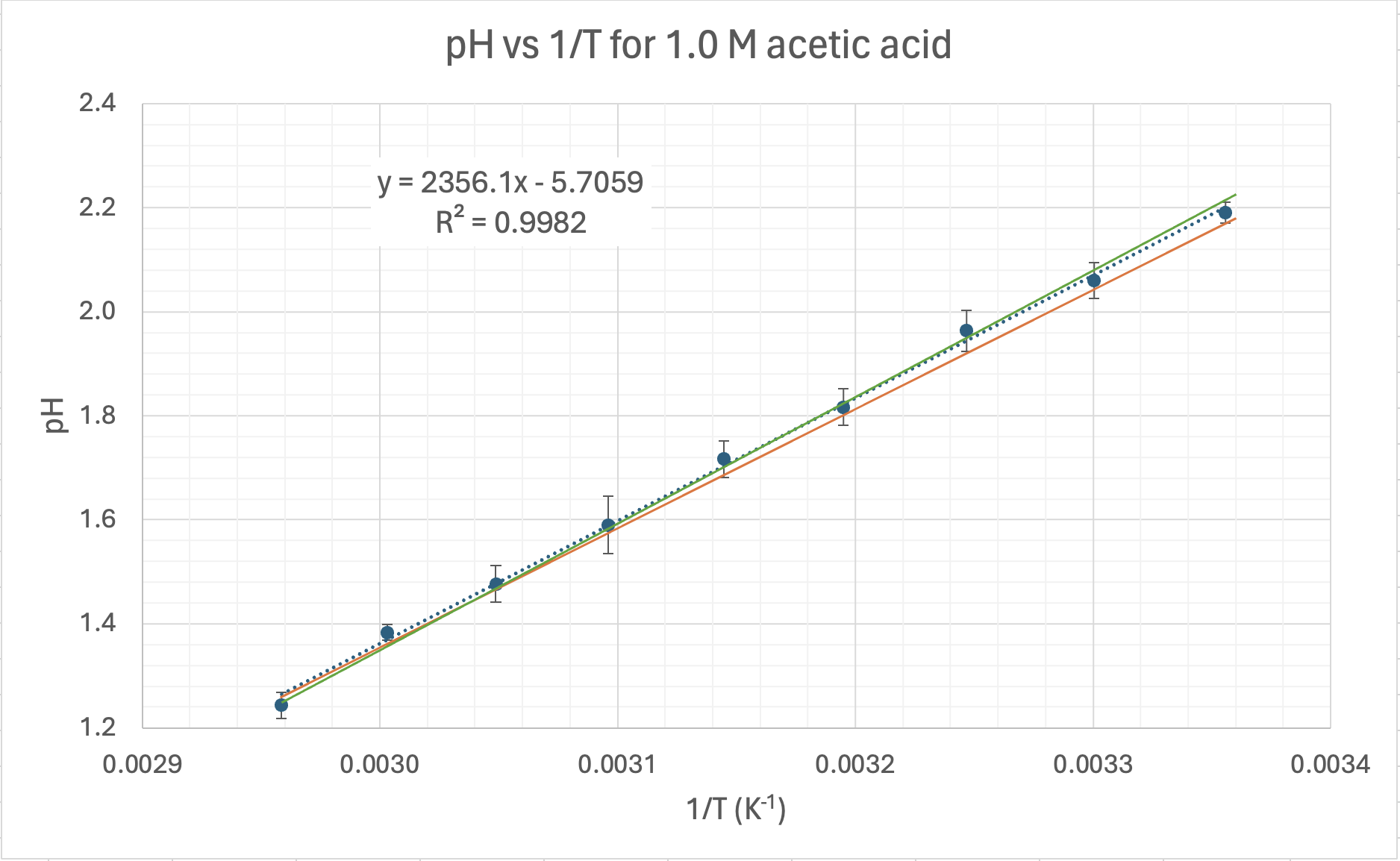








.jpg)